Оглавление
Ключевые слова
- Болезнь малых ножек подоцита;
- болезнь минимальных изменений;
- нефротический синдром;
- стероидзависимый нефротический синдром;
- стероидрезистентный нефротический синдром;
- стероидчувствительный нефротический синдром;
Список сокращений
БМИ – болезнь минимальных изменений
БРА – блокаторы рецепторов ангиотензина
ГН – гломерулонефрит
СЗ СЧНС – стероидзависимая форма стероид-чувствительного нефротического синдрома
и-АПФ – ингибиторы ангиотензинпревращающего фермента
КНИ – кальцинейрина ингибиторы
ММФ – микофенолат мофетил
МК – микофеноловая кислота
МП – метилпреднизолон
НС – нефротический синдром
ОРВИ – острая респираторная вирусная инфекция
РААС – ренин-ангиотензин-альдостероновая система
рСКФ – расчетная скорость клубочковой фильтрации
СРНС – стероидрезистентный нефротический синдром
ФСГС – фокально-сегментаоный гломерулосклероз
ЧР – часто-рецидивирующий
NPHS1 – ген нефрина
NPHS2 – ген подоцина
PLCE1 – фосфолипаза С эпсилон 1
TRPC-6 – потенциал- зависимый временный рецептор катионов 6
NEPH1 – нефриноподобный белок 1
CD2AP – CD2-ассоциированный протеин
ZO-1 – белок плотных контактов (zonula occludens 1)
WT-1 – белок опухоли Вильмса 1
LMX1B – LIM гомеобокс фактор транскрипции 1бета (LIM homeobox transcription factor 1, beta)
SMARCAL1 – подобный, ассоциированный с матриксом; актин- зависимый регулятор хроматина, белок 1 подсемейства альфа
INF2 – инвертированный формин 2.
Термины и определения
Ангиотензин (ангио- + лат. Tensio напряжение; син.: ангиотонин, гипертензин) — биологически активный полипептид, образующийся из ангиотензиногена, повышающий артериальное давление в результате сужения кровеносных сосудов.
Ангиотензин I — неактивная форма а., представляющая собой декапептид, образующийся из ангиотензиногена под действием ренина; предшественник ангиотензина II.
Ангиотензин II — активная форма а., представляющая собой октапептид, образующийся из ангиотензина I под действием пептидазы.
Ангиотензиноген (ангиотензин + греч. -genes порождающий; син. Гипертензиноген) — сывороточный глобулин, образующийся в печени и являющийся предшественником ангиотензина.
Биопсия — микроскопическое исследование прижизненно иссеченных или изъятых другим способом тканей и органов с диагностической целью.
Биоптат — материал, полученный путем биопсии.
Гиперкортицизм – синдром, обусловленный избыточным содержанием в крови кортикостероидов.
Гиперхолестеринемия (hypercholesterinaemia; гипер- + холестерин + греч. Haima кровь; син. Холестеринемия) — повышенное содержание холестерина в крови.
Гипоальбуминемия (hypoalbuminaemia; гипо- + альбумин + греч. Haima кровь) — уменьшенное содержание альбуминов в сыворотке крови; наблюдается при поражениях паренхимы печени, нефротическом синдроме и т. Д.
Гиповолемия (oligaemia; олиг- + греч. Haima кровь) — уменьшенное общее количество крови.
Гипопротеинемия (hypoproteinaemia; гипо- + протеинемия) — пониженное содержание белка в сыворотке крови, наблюдается при его недостаточном поступлении в организм или значительных потерях.
Гломерула (glomerula)- клубочек, часть функциональной единицы почек-нефрона, ответственный за фильтрационную функцию почек.
Гломерулонефрит (glomerulonephritis; гломеруло- + нефрит; син. Брайтова болезнь — устар.) — двустороннее диффузное воспаление почек с преимущественным поражением клубочков.
Гломерулопатия — состояние, при котором отмечается патологические изменения в клубочковом аппарате почек любого генеза
Денситометрия (денсито- + греч. Metreo измерять, определять) —измерение оптической плотности фотопластинки или фотопленки, слоя геля, бумаги и т. Д.; используется, напр., при анализе рентгено- и хроматограмм.
Кушинга синдром (н. W. Cushing; син. Иценко — кушинга синдром) — сочетание характерных изменений внешнего вида больного (ожирение с преимущественным отложением жира на животе и задней части шеи, лунообразное яйцо, гирсутизм, наличие атрофических полос на коже) с артериальной гипертензией, остеопорозом, мышечной слабостью, снижением переносимости глюкозы, у женщин — также с нарушениями менструального цикла; наблюдается при гиперфункции коры надпочечников (чаще при наличии гормонально-активной опухоли), а также при длительном лечении препаратами адренокортикотропного или кортикостероидных гормонов.
Нефротический синдром (НС) — клинико-лабораторный симптомокомплекс, характеризующийся протеинурией, гипоальбуминемией , диспротеинемией, гиперлипидемией, отеками, в том числе полостными.
Протеинурия (proteinuria; протеины + греч. uron моча; син. альбуминурия — устар.) — повышенное содержание белка в моче.
Подоцит — видоизмененный эпителий в структуре клубочкового аппарата почек.
Подоцитопатия — состояние, характеризующееся видоизменением структуры подоцита, обусловленное различными механизмами(иммунными и неиммунными).
Стероидчувствительный НС — наличие эффективности стероидной терапии с достижением полной клинико-лабораторной ремиссии.
Стероидрезистентный НС — отсутствие эффективности стероидной терапии в дозе 60 мг/м2/сут (2 мг/кг/сут) в течение 8 недель, или в дозе 60 мг/м2/сут (2 мг/кг/сут) в течение 6 недель и трех последовательных пульсов метилпреднизолона в дозе 1000 мг/1,73м2 при разовом введении.
Стероидзависисмый НС — развитие рецидива НС при снижении дозы преднизолона или в течение 2 недель после отмены преднизолона.
Фильтрация клубочковая (син. фильтрация гломерулярная) — совокупность процессов перехода веществ, содержащихся в крови, через стенку капилляра клубочка почки в полость его капсулы, что приводит к образованию первичной мочи.
1. Краткая информация
1.1 Определение
Болезнь минимальных изменений (БМИ) – это непролиферативная гломерулопатия, не имеющая каких-либо морфологических критериев при световой микроскопии, обусловленная повреждением (иммунным или неиммунным) подоцитов (подоцитопатия), которое диагностируется исключительно при ультраструктурном анализе в виде диффузного слияния ножковых отростков подоцитов. Повреждение подоцита определяет формирование в клинике заболевания нефротического синдрома (НС).
1.2 Этиология и патогенез
Определенного этиологического фактора при болезни минимальных изменений нет. Однако при многих состояниях может возникнуть нефротический синдром с минимальными изменениями (см. табл.1)
Таблица 1
Состояния, ассоциированные с БМИ
Аллергия:
Пыльца
Грибы
Коровье молоко
Домашняя пыль
Укусы пчел, медуз
Шерсть кошки
Лекарственные препараты:
Нестероидные противовоспалительные препараты
Ампициллин
Препараты золота
Препараты лития
Ртуть-содержащие препараты
Триметадион
Злокачественные заболевания:
Болезнь Ходжкина
Неходжкинская лимфома
Рак толстой кишки
Карцинома легких
Другие:
Вирусная инфекция
Болезнь Кимуры
Сахарный диабет
Миастения Грависа
Вакцинация
В патогенезе БМИ следует рассматривать два механизма развития:
Иммуноопосредованный:
В настоящее время исследования in vivo и in vitro продемонстрировали высокую активность Т-лимфоцитов в ответ на антигенную стимуляцию. В последующем происходит дифференцировка Т-клеток с преимущественным образованием Тh2, экспрессирующих IL-4 и IL-13. Более того активация NFkB транскрипционного фактора наблюдается при всех случаях рецидивов НС при БМИ. Антагонистом NFkB является IkB, концентрация которого под влиянием глюкокортикоидов увеличивается. Эффективность ритуксимаба при лечении БМИ предполагает роль В-клеток в патогенезе БМИ. Одной из гипотез развития протеинурии при БМИ является повреждение щелевой диафрагмы, регулируемое экспрессией CD80 (В7-1) на подоцитах – трансмембранного протеина, экспрессирующегося на антиген-презентирующих клетках (АПК), натуральных киллерах и В-лимфоцитах. CD80 определяет ко-стимулирующий сигнал для Т-лимфоцитов, связываясь с последними посредством соединения с их рецепторами CD28. Данный механизм отмечается при представлении антигена АПК Т-клеткам с последующей их активацией. Однако связывание CD28 на Т-лимфоцитах с CTLA-4 – протеина, экспрессируемого на Foxp3+ регуляторных Т-клетках (Treg) ингибирует активацию последних. Мутация в гене Foxp3 у больных БМИ приводит к снижению активации Treg клеток, тем самым способствуя развитию протеинурии.
Неиммунный:
Структура подоцита изменяется в результате изменения структурных белков подоцитов, обусловленное мутациями генов. До 66% случаев НС на первом году жизни у детей составляет генетически обусловленный НС. Частота генетических форм НС у детей при идиопатическом НС неизвестна. Однако следует помнить морфологический диагноз БМИ у детей с генетически обусловленным НС носит транзиторный характер, так как в последующем он трансформируется в ФСГС. Неиммунный характер формирования БМИ определяет развитие стероид-резистентной формы БМИ.
1.3 Эпидемиология
- БМИ составляет 76,6 % всех морфологических вариантов первичного гломерулонефрита (ГН) у детей.
- Наибольшая встречаемость у детей раннего возраста.
- БМИ чаще отмечается у мальчиков в соотношении 2:1
- Возможны семейные формы, обусловленные мутациями генов структурных белков подоцита.
- Рецидивов в трансплантате нет.
1.4 Коды по МКБ-10
N04.0 — Нефротический синдром с незначительными гломерулярными нарушениями
1.5 Классификация
Официально утвержденной классификации болезни минимальных изменений нет. Однако с учетом этиологического фактора можно разделить данную патологию на две формы:
Первичная (идиопатическая) БМИ
Основой развития идиопатического нефротического синдрома у детей является дисфункция Т-клеточного звена иммунной системы или генетические мутации. Однако БМИ может быть ассоциирована с множеством других патологических состояний таких, как аллергия, онкопатология, лекарственные воздействия.
Генетически обусловленная БМИ (гены):
- Щелевой диафрагмы и цитоскелета подоцитов – NPHS1, NPHS2, TRCP6, CD2AP, ACTN4, INF2;
- Фосфолипазы – PLCE1;
- Гломерулярной базальной мембраны – LAMB2;
- Факторов транскрипции – WT1, LMX1B;
- Лизосомных белков – SCARB2;
- Митохондриальных белков – COQ2;
- Посредника реструктуризации ДНК-нуклеосомы — SMARCAL1.
2. Диагностика
Дифференциальную диагностику проводят с другими формами ГН, дебютировавшими НС (НГ). Дифференциальная диагностика проводится в случае развития стероидзависимой и стероидрезистентной формы НС. (1В)
Клинические проявления БМИ не различаются при идиопатическом и вторичном вариантах заболевания. В этой связи дифференциальная диагностика этих форм должна базироваться на исключении всех возможных вторичных причин БМИ (см. классификацию) (НГ).
Детальное морфологическое исследование ткани почки, включающее световую, иммуногистохимическую и электронную микроскопию, обязательно для диагностики БМИ (НГ).
Морфологические критерии БМИ
Световая микроскопия:
На светооптическом уровне – при БМИ клубочек выглядит не поврежденным, иногда может присутствовать минимальная мезангиальная пролиферация (до 3-х клеток), что создает трудности в дифференциации с минимальными изменениями при мезангиопролиферативном гломерулонефрите. У детей с часторецидивирующим БМИ некоторые клубочки могут быть инволютированы.
Клетки канальцев инфильтрированы белками и липидами из-за увеличенной реабсорбции. Наличие атрофии и фиброза канальцев должны вызвать подозрение на наличие фокально-сегментарного гломерулосклероза.
Иммуногостохимия:
При иммуногистохимическом исследовании отмечается отсутствие отложения иммуноглобулионов и компонентов комплемента.
Электронная микроскопия:
Диффузное «сглаживание» ножек подоцитов является гистологическим маркером БМИ при сочетании с вышеизложенной световой микроскопией и иммуногистохимическим исследованием.
2.1 Жалобы и анамнез
Диагноз БМИ устанавливается на основании клинико-лабораторной картины НС и быстрому положительному ответу на стероидную терапию (НГ). Морфологическая диагностика является резервным методом при атипичном клиническом ответе на терапию.
- Клиническая диагностика идиопатической БМИ должна базироваться на развитии НС у детей раннего и дошкольного возраста (HГ).
- Наличие в анамнезе состояний, ассоциированных с БМИ (табл. 1) и ранний возраст дебюта НС следует рассматривать как факторы, определяющие развитие БМИ.
- Развитие НС на первом году жизни и в подростковом возрасте должны насторожить врача в пользу генетически обусловленного НС или другой морфологической формы НС.
- Клиническим синдромом БМИ является внезапно развившийся НС (протеинурия, гипоальбуминурия, гиперлипидемия). Отягощенный аллергологический анамнез и аллергические проявления у детей с БМИ наблюдаются в 30-70% случаев в отличие от других форм гломерулонефрита. Триггерными факторами могут быть ОРВИ, детские инфекции, атопические реакции (см. выше табл.1).
2.2 Физикальное обследование
Артериальная гипертензия наблюдается крайне редко и характеризуется кратковременностью. Повышение артериального давления при БМИ связано с компенсаторным механизмом на выраженную гиповолемию. При резкой гиповолемии возможно развитие нефротического криза с болями в животе, кожной эритемой и сердечно-сосудистым шоком с циркуляторной недостаточностью.
- Рекомендовано обратить внимание на отеки.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Комментарии: Первым клиническим симптомом, заметным для больного и окружающих, являются отеки. Они могут развиться постепенно или же стремительно, достигнув степени анасарки. Периферические отеки выявляются в области век, лица, поясничной области и половых органов, могут распространяться на всю подкожную клетчатку, растягивая кожу до образования стрий. В это время у больных могут образовываться транссудаты в серозные полости: одно- или двусторонний гидроторакс, асцит, гидроперикард; возможно развитие отека легких.
2.3 Лабораторная диагностика
- Рекомендовано проведение клинического анализа крови и определение гематокритного показателя.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Комментарии:
- Возможна умеренная анемия;
- Повышенный гематокрит (> 44%);
- Тромбоцитоз;
- Вторичный лейкоцитоз на фоне приема глюкокортикостероидов.
- Выраженное повышение СОЭ.
- Рекомендовано проведение биохимического анализа крови.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Комментарии:
- Гипопротеинемия (<55г/л);
- Гиопаальбуминемия(<25г/л);
- Гиперхолестеринемия (>5,7ммоль/л);
- Диспротеинемия (повышение ?2-фракций глобулинов и снижение ?-глобулинов).
- Рекомендовано проведение общего анализа мочи.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Комментарии:
-
- Выраженная протеинурия (> 3г/л);
- Редко микрогематурия до 10 эр. в п /зрения;
- Цилиндрурия (гиалиновые).
- Рекомендовано определение суточной экскреции белка для уточнения степени протеинурии.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Комментарии: Протеинурия> 1г/м2/сутки или >40 мг/м2/сут. При невозможности определения суточной экскреции белка для уточнения степени протеинурии может быть использовано определение отношения уровня экскретируемого белка к креатинину в разовой порции мочи. Этот коэффициент достоверно коррелирует с уровнем суточной протеинурии/1,73м2 Экскреция белка (г/сут/1,73м2) = (белок г/л*0,088) /креатинин мочи (ммоль/л)
- Рекомендовано проведение коагулограммы.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Комментарии:
- Гиперпротромбинемия;
- Гиперфибриногенемия;
- Повышения D-димеров;
- Снижение антитромбина III.
- Рекомендованы иммунологические исследования.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
Комментарии: Возможно повышение IgE, Низкий уровень IgG.
- Рекомендованы определение СКФ методом Шварца.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
- Рекомендованы проба Реберга для оценки клиренса эндогенного креатинина.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
Комментарии: Возможно снижение клубочковой фильтрации и клиренса эндогенного креатинина в активную стадию на фоне выраженной гиповолемии.
- Рекомендовано генетическое (гены см. выше) исследование в случае, если НС наблюдается до 1 года и при стероидрезистентной форме.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
2.4 Инструментальная диагностика
- Рекомедовано проведение ЭКГ- электрографические признаки гидроперикарда.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
- Рекомедовано проведение Эхо-ЭКГ — эхографические признаки гидроперикарда.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
- Рекомендовано проведение ультразвукового исследования почек.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
Комментарии: Увеличение размеров почек, гипоэхогенность коркового слоя;
- Рекомендовано денситометрия поясничного отдела позвоночника или рентгенография трубчатых костей для оценки степени деминерализации костной ткани;
Уровень убедительности рекомендаций С (уровень достоверности – 4)
- Рекомендована пункционная биопсия почек (по показаниям) в случае, если дебют заболевания раннее 1 года и старше 12 лет, стероидрезистентная форма.
Уровень убедительности рекомендаций С (уровень достоверности – 4)
3. Лечение
Показания к госпитализации:
- Все дети в активную стадию должны быть госпитализированы в стационар. Длительность пребывания в стационаре в среднем 14-21 день.
Дети в стадии ремиссии могут наблюдаться в амбулаторных условиях.
3.1 Лечение первого эпизода стероидчувствительного нс при БМИ (НГ)
При развитии НС у детей до года до начала кортикостероидной терапии следует проводить нефробиопсию.
Немедикаментозное лечение
- Не рекомендовано ограничение двигательной активности.
Уровень убедительности рекомендаций С (уровень достоверности –4)
- Рекомендована сбалансированная диета, количество потребляемого белка- 1,5-2 г/кг и сохранение калоража пищи за счет полиненасыщенных жиров. Бессолевая или с низким содержанием соли (<2гNa / день) только в период выраженных отеков. При тяжелых отеках: ограничение потребления жидкости.
Уровень убедительности рекомендаций С (уровень достоверности –4)
Медикаментозное лечение
- Рекомендовано назначать кортикостероидную терапию (преднизолон) на период не менее 12 недель.
Уровень убедительности рекомендаций B (уровень достоверности –1)
- Рекомендовано применять преднизолон внутрь ежедневно в 1 или 2 приема (1B) в начальной дозе 60 мг/м2/24 ч или 2 мг/кг/24 ч, максимально до 60 мг/24 ч (1D) в течение 4–6 недель (1C) с последующим переходом на прием препарата через день (альтернирующий прием), начиная с дозы 40 мг/м2 или 1,5 мг/кг (максимум 40 мг через день) в один прием (1D) с постепенным снижением дозы в течение 2–3 мес. (1B).
Уровень убедительности рекомендаций B-D (уровень достоверности –1)
Комментарии: Общая длительность терапии должна составлять 4-5мес (1B).
3.2 Лечение рецидивирующей формы НС при БМИ
Кортикостероидная терапия у детей с редкими рецидивами стероидчувствительного НС при БМИ.
- У детей с редкими рецидивами стероидчувствительного нефротического синдрома (СЧНС) рекомендовано проводить лечение преднизолоном в дозе 60 мг/м2 или 2 мг/кг (максимально 60 мг/24 ч) в 1 или 2 приема до тех пор, пока не будет констатирована полная ремиссия в течение 3 дней.
Уровень убедительности рекомендаций D (уровень достоверности –2)
- После достижения ремиссии рекомендовано назначается преднизолон в дозе 40 мг/м2 или 1,5 мг/кг (максимально 40 мг) через день в течение как минимум 4 недель.
Уровень убедительности рекомендаций С (уровень достоверности –2)
Кортикостероидная терапия у детей с часто рецидивирующими и стероидзависимыми формами стероидчувствительного НС при БМИ.
- При рецидивах часто рецидивирующей (ЧР) и стероидзависимой формы (СЗ) СЧНС рекомендовано назначать преднизолон ежедневно до тех пор, пока полная ремиссия не будет констатирована в течение не менее 3 дней, и затем преднизолон в альтернирующем режиме в течение не менее чем 3 мес.
Уровень убедительности рекомендаций С (уровень достоверности –2)
- У детей с ЧР и СЗ СЧНС рекомедовано рассмотреть возможность назначение преднизолона в альтернирующем режиме в самых низких дозах, необходимых для поддержания ремиссии, во избежание серьезных побочных эффектов. В случае неэффективности данной схемы возможен ежедневный прием в минимальной суточной дозе, необходимой для поддержания ремиссии без серьезных побочных эффектов.
Уровень убедительности рекомендаций D (уровень достоверности –2)
- У детей с ЧР и СЗ СЧНС, получающих преднизолон в режиме через день, на период эпизодов респираторных и других инфекций рекомендовано назначать преднизолон ежедневно, с целью уменьшения риска обострений.
Уровень убедительности рекомендаций С (уровень достоверности –2)
3.3 Лечение ЧР и СЗ СЧНС кортикостероидсберегающими препаратами
Алкилирующие препараты при лечении часто рецидивирующей и стероидзависимой формы стероидчувствительного НС при БМИ. Эффективность составляет от 30% до 50%.Основные осложнения терапии: цитопения, инфекционные поражения, токсический гепатит, геморрагический цистит, гонадотоксичность.
Показаниями у биопсии почки у детей с СЧНС являются (НГ):
- отсутствие эффекта при рецидивах после первоначального ответа на кортикостероиды;
- высокий индекс подозрения в отношении иной основной патологии;
- ухудшение функции почек у детей, получающих КНИ.
- Рекомендовано назначать стероидсберегающие препараты у детей с ЧР и СЗ СЧНС в тех случаях, когда развиваются побочные эффекты кортикостероидной терапии.
Уровень убедительности рекомендаций В (уровень достоверности –1)
- При ЧР (1B) и СЗ (2C) СЧНС рекомендовано использовать в качестве стероидсберегающих препаратов алкилирующие агенты — циклофосфамид или хлорамбуцил.
Уровень убедительности рекомендаций С (уровень достоверности –2)
- Назначать циклофосфамид в дозе 2 мг/кг/24 ч в течение 8–12 нед (максимальная кумулятивная доза 168 мг/кг).
Уровень убедительности рекомендаций С (уровень достоверности –2)
- Не рекомендовано начинать терапию циклофосфамидом до тех пор, пока не будет достигнута ремиссия с помощью кортикостероидов (2D).
Уровень убедительности рекомендаций D (уровень достоверности –2)
- Рекомедовано назначать хлорамбуцил в дозе 0,1–0,2 мг/кг/24 ч в течение 8 нед (максимальная кумулятивная доза 11,2 мг/кг) в качестве альтернативы циклофосфамиду.
Уровень убедительности рекомендаций С (уровень достоверности –2)
- На фоне приема алкилирующих препаратов глюкокортикостероидная терапия должна быть окончена не более чем за 2 недели до окончания курса алкилирующих препаратов.
Уровень убедительности рекомендаций С (уровень достоверности –2)
- Не рекомендовано проводить второй курс алкилирующих препаратов.
Уровень убедительности рекомендаций D (уровень достоверности –2)
Левамизол при лечении часто рецидивирующей и стероидзависимой формы стероидчувствительного НС при БМИ
- При лечении ЧР и СЗ СЧНС (1В) рекомедовано назначить левамизол в дозе 2,5 мг/кг через день (2B) в течение как минимум 12 мес (2C), так как у большинства детей при отмене левамизола возникают рецидивы. Препарат назначать под контролем уровня нейтрофилов.
Уровень убедительности рекомендаций В (уровень достоверности –2)
Ингибиторы кальцинейрина (циклоспорин или такролимус) при лечении часто рецидивирующей и стероидзависимой формы стероидчувствительного НС при БМИ.
- Рекомендовано назначать КНИ в течение как минимум 12 мес, так как у большинства детей при отмене КНИ развиваются обострения.
Уровень убедительности рекомендаций С (уровень достоверности –2)
- Рекомендовано применять циклоспорин А в начальной дозе 4–6 мг/ кг/24 ч в 2 приема.
Уровень убедительности рекомендаций С (уровень достоверности –2)
Комментарии: Начало терапии при достижении ремиссии на фоне глюкокортикоидной терапии и переходе на альтернирующий режим. Контроль эффективности дозы осуществляется путем измерения концентрации препарата в сыворотке крови. Определение концентрации циклоспорина А возможно в двух точках: в точке С0 – определение базального уровня циклоспорина до утреннего приема препарата(или через 12 часов от вечернего приема); в точке C2— определение концентрации через 2 часа после утреннего приема препарата. Эффективная концентрация циклоспорина А при ЧР и СЗ СЧНС при БМИ следующая:
С0— 80-100нг/мл
С2 – 700-800нг/мл
Эффективность терапии- 80-90%.
- Рекомендовано применять такролимус в начальной дозе 0,1 мг/кг/24 ч в 2 приема вместо циклоспорина А в случае выраженных косметических побочных эффектов циклоспорина.
Уровень убедительности рекомендаций D (уровень достоверности –2)
Комментарии: Принцип назначения такролимус такой как циклоспорина А, т.е. контроль эффективности дозы определяется базальным уровнем концентрации препарата в сыворотке крови.
Эффективная концентрация такролимуса в т. С0 – 5-8 нг/мл.
Эффективность терапии- 60-80%.
Основные осложнения терапии: нефротоксичность. При снижении скорости клубочковой фильтрации (СКФ) на 30% дозу КНИ уменьшают вдвое, при снижении СКФ на 50% — отменяют препарат. При длительности терапии более 2,5-3 лет рекомендуется проведение нефробиопсии для выявления возможных морфологических признаков токсичности (повреждение эпителия канальцев, склероз интерстиция и стенок артериол). Также среди побочных действий ЦСА – гепатотоксичность, гиперурикемия, гипертрихоз, гиперкалиемия, гипомагнезиемия, гиперплазия десен.
- Для уменьшения токсичности следует мониторировать концентрацию ингибиторов кальцинейрина в сыворотки крови (КНИ).
Уровень убедительности рекомендаций НГ
- Рекомендовано назначать КНИ в течение как минимум 12 мес, так как у большинства детей при отмене КНИ развиваются обострения.
Уровень убедительности рекомендаций С (уровень достоверности – 2)
Микофенолаты при лечении часто рецидивирующей и стероидзависимой формы стероидчувствительного НС при БМИ
- Рекомендовано назначать микофенолат мофетил в начальной дозе 1200 мг/м2/24 ч или микофеноловую кислоту начальной дозе 720 мг/м2 в 2 приема в течение как минимум 12 мес, так как у большинства детей при отмене микофенолатов развиваются рецидивы (2C).
Уровень убедительности рекомендаций С (уровень достоверности – 2)
Комментарии: Эффективность терапии составляет 50-60%.
Ритуксимаб при лечении часто рецидивирующей и стероидзависимой формы стероидчувствительного НС при БМИ.
- Рекомендовано применять ритуксимаб только у тех детей со СЗ СЧНС, у которых частые рецидивы возникают несмотря на применение оптимальых комбинаций преднизолона и кортикостероидсберегающих препаратов или у тех, у которых развиваются серьезные побочные эффекты этой терапии.
Уровень убедительности рекомендаций С (уровень достоверности – 2)
Комментарии: Введение препарата возможно только в условиях стационара в дозе 375мг/2 внутривенно с еженедельным введением в течение 4 недель.
- Не рекомендовано использовать мизорибин? в качестве кортикостероидсберегающего препарата при ЧР и СЗ СЧНС при БМИ.
Уровень убедительности рекомендаций С (уровень достоверности – 2)
- Не рекомендовано использовать азатиоприн в качестве кортикосте-роидсберегающего препарата при ЧР и СЗ СЧНС при БМИ.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
3.4 Лечение стероидрезистентной формы НС при БМИ
Для оценки детей со СРНС требуется (НГ):
- диагностическая биопсия почки;
- оценка функции почек по СКФ и рСКФ;
- количественная оценка экскреции белка.
- Рекомендовано констатировать стероидную резистентность после 8 недель стероидной терапии без эффекта или 3 пульсов-терапии метил преднизолоном в дозе 20-30мг/кг, но не более 1г/сут. после 6 недель.
Уровень убедительности рекомендаций D (уровень достоверности – 2)
- Рекомендовано использовать КНИ в качестве инициальной терапии у детей со СРНС.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
- Рекомендовано роводить терапию КНИ в течение как минимум 6 мес. и прекращать ее, если к этому времени не достигнута частичная или полная ремиссия ПУ.
Уровень убедительности рекомендаций С(уровень достоверности – 2)
- Рекомендовано продолжить терапию КНИ в течение как минимум 12 мес., если через 6 мес. достигнута хотя бы частичная ремиссия (2C).
Уровень убедительности рекомендаций С (уровень достоверности – 2)
Комментарии: Эффективная доза КНИ определяется определением их концентраций в сыворотке крови.
При СРНС эффективная лечебная концентрация циклоспорина А и такролимуса составляют:
ЦисА:
т. С0— 100-120нг/мл
т. С2— 1000-1200нг/мл
Так.:
т.С0— 6-8нг/мл соответственно
- Рекомендовано комбинировать терапию малыми дозами кортикостероидов с терапией КНИ.
Уровень убедительности рекомендаций D (уровень достоверности – 2)
- Всем детям со СРНС рекомендовано проводить лечение иАПФ или БРА детям со СРНС как в качестве гипотензивной, так и в качестве нефропротективной терапии.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
- При высокой активности СРНС рекомендовано использовать пульс-терапию метилпреднизолоном (МП) в сочетании с КНИ: схема Вальдо (табл. 2).
Уровень убедительности рекомендаций В (уровень достоверности – 1)
Комментарии: Таблица 2 — Схема Вальдо
|
Неделя |
МП 30 мг/кг в/в |
Преднизолон |
Циклоспорин А |
|
1-2 |
3 раза в неделю |
— |
— |
|
3-8 |
1 раз в неделю |
2 мг/кг через день |
6 мг/кг/24ч |
|
9-29 |
— |
1 мг/кг через день |
3 мг/кг/24ч |
|
30-54 |
— |
0,5 мг/кг через день |
3 мг/кг/24ч |
У детей, не достигших ремиссии на терапии КНИ:
- Рекомендовано использовать микофенолата мофетил и высокие дозы кортикостероидов или комбинацию этих препаратов у детей, не достигших полной или частичной ремиссии на КНИ и кортикостероидах.
Уровень убедительности рекомендаций D (уровень достоверности – 2)
- Не рекомендовано назначать циклофосфамид детям со СРНС.
Уровень убедительности рекомендаций В (уровень достоверности – 2)
- У пациентов с рецидивом нефротического синдрома после достижения полной ремиссии возобновить терапию с использованием одной из следующих схем:
- кортикостероиды внутрь;
- вернуться к тому иммуносупрессивному препарату, который ранее был эффективен;
- использовать альтернативный иммуноспурессивный препарат для уменьшения кумулятивной токсичности.
Уровень убедительности рекомендаций D (уровень достоверности – 2)
3.6 Симптоматическая терапия
- Рекомендована диуретическая терапия для лечения больных с отеками.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
Комментарии: Диуретическая терапия широко используются для лечения больных с отеками:
- Гидрохлоротиазид: 2 -4 мг / кг/сут;
- Верошпирон: 2 ~ 4 мг /кг;
- В/в декстраны: 10 ~ 15мл/кг, с последующим введением фурасемида (Лазикс) 2-4мг/кг, спустя 30 ~ 60мин;
- В/в альбумин (20%-до 5 мл/кг) +лазикс;
Показания для в/в альбумина:
- Тяжелые отеки;
- Асцит;
- Гидроторакс и гидроперикард;
- Генитальные отеки;
- Низкий уровень альбумина ( <20г/л).
3.7 Лечение осложнений:
Гипертония:
- Рекомендовано назначать с гипотензивной и нефропротективной целью ингибиторы ангиотензин-превращающего фермента (иАПФ): фозиноприл или эналаприл индивидуальный подбор дозы, в среднем: 0,1- 0,3 мг/кг по фозиноприлу и блокаторы рецепторов ангиотензина (БРА).
Уровень убедительности рекомендаций В (уровень достоверности – 1)
- Рекомендовано применять И АПФ и БРА при отсутствии эффекта от ранее проводимых всех видов иммуносупрессивной терапии.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
Гиперкоагуляция:
- Рекомендовано проводить антикоагулянтную терапию с целью профилактики венозных и артериальных тромбозов. проводится в активную стадию заболевания под контролем коагулограммы.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
Комментарии: В условиях стационара лучше использовать антикоагулянты с коротким периодам выведения для дальнейшей быстрой коррекции: гепарин в суточной дозе 150-200 Ед/кг/сутки подкожно в 4 приема или фраксипарин 170 МЕ/кг/сут подкожно 1 раз в сутки. Антикоагулянтная терапия проводиться под контролем коагулограммы. При стабилизации дозу гепарина снижают (начинают со снижения дозы, а затем кратности введения).Антиагреганты-дипиридамол (курантил) в дозе 5-8 мг/кг/сутки или тиклодипин (тиклид) 8 мг/кг/сутки, у подростков возможно использование клопидогрела (плавикса) в дозе 75 мг 1 раз в сутки.
Коррекция остеопении и остеопороза:
- Рекомендован витамин D3 в дозе 1000-3000 МЕ в сутки в сочетании с препаратами кальция. 1000–1500 мг/сут (по элементарному кальцию).
Уровень убедительности рекомендаций В (уровень достоверности – 1)
Профилактика язвенной болезни
- На фоне приема лечебной дозы глюкокортикостероидов с целью профилактики язвенной болезни рекомендовано назначать ингибиторы протонной помпы или блокаторы Н2— гистаминовых рецепторов в возрастной дозе.
Уровень убедительности рекомендаций В (уровень достоверности – 1)
4. Реабилитация
Реабилитация больных БМИ не проводится.
5. Профилактика и диспансерное наблюдение
5.1. Профилактика
5.1.1 Первичная профилактика не проводится.
5.1.2 Профилактика обострения заболевания
У больных с установленным БМИ в эпидемический период проводится профилактика ОРВИ с использованием немедикаментозных и медикаментозных методов профилактики (НГ).
- В случае развития ОРВИ у больного с БМИ на фоне приема иммуносупрессивных препаратов с целью профилактики обострения процесса рекомендовано назначать антибактериальную терапию.
Уровень убедительности рекомендаций С (уровень достоверности –4)
- Рекомендация 42. Для уменьшения риска серьезных инфекций у детей с СЧ БМИ следует (НГ):
- проводить детям противопневмококковую вакцинацию.
- проводить вакцинацию против гриппа ежегодно детям, и всем, кто проживает с ними совместно.
- отложить вакцинацию живыми вакцинами до тех пор, пока доза преднизолона не будет снижена до 1 мг/кг/24ч (<20 мг/24ч) или до 2 мг/кг через день (<40 мг через день).
- живые вакцины противопоказаны детям, получающим кортикостероид-сберегающие иммуносупрессивные препараты.
- для уменьшения риска инфицирования детей с подавленными иммунитетом иммунизировать здоровых лиц, проживающих совместно с детьми живыми вакцинами, но обеспечить отсутствие контакта детей с выделениями мочевой, пищеварительной и дыхательной систем вакцинированных лиц в течение 3-6 недель после вакцинации.
- при контакте с ветряной оспой – не привитым детям, получающим иммуносупрессанты, при возможности назначать противозостерный иммуноглобулин.
Уровень убедительности рекомендаций С (уровень достоверности –4)
5.2 Диспансерное наблюдение
- Длительность наблюдения не менее 5 лет(2С).
- Наблюдение проводится участковым педиатром и нефрологом. Частота осмотра представлена в таблице 3.
- В комплекс диспансерного наблюдения входят определения режима, диеты, санаторно-курортное лечение.
- Диета у больного с БМИ должна быть гипоаллергенной, с исключением экстративных веществ, и сбалансированная по калоражу согласно возрасту.
- Режим – ограничений двигательной активности нет.
- Обязательное проведение санации очагов инфекции, с этой целью проводится осмотр стоматологом и отолорингологом. Частота осмотра представлена в табл. 3
- В перечень лабораторных исследований в период диспансерного наблюдения у больного с БМИ входят: общий анализ мочи, клинический анализ крови, определение суточной экскреции белка, количественный анализ мочи (Амбурже или Нечипоренко), проба Зимницкого, биохимический анализ крови, функциональная с определением СКФ или клиренса эндогенного креатинина. Частота исследований представлены в таблице 3.
- Снятие с учета через 5 лет полной ремиссии после обследования в нефрологическом стационаре, стационаре одного дня, диагностическом центре.
Таблица 4. Примерная схема диспансерного наблюдения детей с острым гломерулонефритом (по М.В. Эрману, 1997)
|
Частота осмотров специалистами |
Дополнительные методы обследования |
Основные пути оздоровления |
|
· Педиатр 1-ый год: В первые 3 мес. — 2 раз/месяц С 3 до 12 мес. — 1 раз/месяц Затем 1 раз в 2-3 мес.
1-ый год: 1 раз в 3 мес. Затем 1-2 раза в год
1 раз в 6 мес.
|
1. Анализ мочи Первые 6 мес. — 1раз в 10-14 дней, затем 1 раз в мес.
|
Режим Диета Реабилитация в местном нефрологическом санатории При интеркуррентных заболеваниях симптоматическая терапия. Анализы мочи при заболевании, выздоровлении через 2-3 мес. Медицинский отвод от прививок на го |
Критерии оценки качества медицинской помощи
|
№ |
Критерии качества |
Уровень достоверности доказательств |
Уровень убедительности рекомендаций |
|
1 |
Выполнено определение уровня суточной протеинурии и оценка биохимических показателей НС (уровень альбумина сыворотки крови, уровень холестерина сыворотки крови |
А |
1 |
|
2 |
Выполнена оценка скорости клубочковой фильтрации |
В |
1 |
|
3 |
Выполнено ультразвуковое исследование почек |
С |
4 |
|
4 |
Выполнен общий анализ мочи с микроскопией мочевого осадка |
С |
4 |
Список литературы
- Детская нефрология. /Под ред. Н. Сигела /Пер.А. Александровского, Д. Буйнова, А. Вермеля, А. Засядько, Д. Колода, Е. Макаренко, А. Мишарина, Ю. Ольшанской, А. Рылова, Н. Первухова. М.: Практика 2006; 336.
- Детская нефрология. /Под ред. Э. Лойманна, А.Н. Цыгина, А.А. Саркисяна. М.: Литтерра — 2010.
- Детская нефрология Руководство для врачей. /Под ред. М.С. Игнатовой, 3-е изд. М.: МИА 2011;696.
- Диагностика и лечение нефротического синдрома у детей: Руководство для врачей. М.С. Игнатова, О.В. Шатохина. М.: МИА 2009; 300.
- Клиническая нефрология.
http://www.sma.org.sg/handheld/express/guidelines/01_06.htm
/Под ред. Папаян А.В., Савенковой Н.Д. С-П.: Сотис 2008; 712.
- Нефрология детского возраста. Руководство для врачей. М.В. Эрман М.: Спецлит 2010; 683.
- Davin J.-C., Rutjes N.W. Nephrotic Syndrome in Children: From Bench to Treatment. International Journal of Nephrology. 2011;8:1-6.
- Dorresteijn E.M., Kist-van Holthe J.E., Levtchenko E.N. et al. Mycophenolate mofetil versus cyclosporine for remission maintenance in nephrotic syndrome. Pediatric Nephrology, 2008; 23(11):2013–2020.
- Eddy A.A., Symons J.M. Nephrotic syndrome in childhood. Lancet 2003; 362(9384):629–639.
- Garin EH, Mu W, Arthur JM, Rivard CJ, Araya CE, Shimada M, et al. Urinary CD80 is elevated in minimal change disease but not in focal segmental glomerulosclerosis. Kidney Int. 2010;78: 296-302.
- Hinkes B.G., Mucha B., Vlangos C.N. et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics, 2007; 119(4): e907–e919.
- Hodson E.M., Willis N.S., Craig J.C. Interventions for idiopathic steroid-resistant nephrotic syndrome in children. Cochrane Database of Systematic Reviews, 2010;11: Article ID CD003594.
- Hodson E.M., Willis N.S., Craig J.C. Non-corticosteroid treatment for nephrotic syndrome in children. Cochrane Database of Systematic Reviews, 2008; 1: Article ID CD002290.
- Ishimoto T, Cara-Fuentes G, Wang H, Shimada M, Wasserfall CH, Winter WE, Rivard CJ, Araya CE, Saleem MA, Mathieson PW, Johnson RJ, Garin EH. Serum from minimal change patients in relapse increases CD80 expression in cultured podocytes. Pediatr Nephrol. 2013 Sep;28(9):1803-1812.
- Ishimoto T, Shimada M, Gabriela G, Kosugi T, Sato W, Lee PY, Lanaspa MA, Rivard C, Maruyama S, Garin EH, Johnson RJ. Toll-like receptor 3 ligand, polyIC, induces proteinuria and glomerular CD80, and increases urinary CD80 in mice. Nephrol Dial Transplant. 2013; 28(6):1439-1446.
- Kimata H., Fujimoto M., Furusho K. Involvement of interleukin (IL)-13, but not IL-4, in spontaneous IgE and IgG4 production in nephrotic syndrome. European Journal of Immunology, 1995; 25(6): 1497–1501.
- Lai K.W., Wei C.L., Tan L.K. et al. Overexpression of interleukin-13 induces minimal-change-like nephropathy in rats Journal of the American Society of Nephrology, 2007; 18(5):1476–1485.
- Machuca E., Benoit G., Antignac C., Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Human Molecular Genetics, 2009;18(R2): R185–R194.
- Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney International. 1978;13(2):159-165.
- Niaudet P. Steroid-sensitive nephrotic syndrome in children in Paediatric Nephrology. /E.D. Avner, W.E. Harmon and P. Neasden, Eds. 2004; 543–556, Lippincott Williams and Wilkins, Philadelphia, Pa, USA.
- Reiser J, Mundel P. Danger signaling by glomerular podocytes defines a novel function of inducible B7-1 in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol. 2004;15:2246-2248.
- Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390-1397.
- Siegel N.J., Gur A., Krassner L.S., Kashgarian M. Minimal-lesion nephrotic syndrome with early resistance to steroid therapy. J Pediatr 1975;87(3):377–380.
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3 regulatory T cell function. Science. 2008; 322: 271-275.
- Yap H.K., Cheung W., Murugasu B.,. Sim S.K, Seah C.C., Jordan S.C. Th1 and Th2 cytokine mRNA profiles in childhood nephrotic syndrome: evidence for increased IL-13 mRNA expression in relapse. Journal of the American Society of Nephrology, 1999;10(3):529–537.
- Yu CC, Fornoni A, Weins A, Hakroush S, Maiguel D, Sageshima J, et al. Abatacept in B7?1?positive proteinuric kidney disease. N Engl J Med 2013;369:2416?2423.
Приложение А1. Состав рабочей группы
Рабочая группа:
- Петросян Э.К. – д.м.н., профессор, нефролог
- Длин. В.В. – д.м.н., профессор, нефролог
Конфликтов интересов: нет
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций являются:
-
-
-
- Врач-педиатр;
- Врач-нефролог.
-
-
-
Для рекомендаций сила указана как уровень 1, 2 или «нет степени» (табл.5), качество доказательной базы обозначено как А, В, С (табл.6).
Таблица 5. Оценка силы рекомендаций (составлена в соответствии с клиническими рекомендациями KDIGO).
Уровень
Оценка рекомендаций
Со стороны пациентов
Со стороны врача
Дальнейшее направление использования
Уровень 1 «Эксперты рекомендуют»
Подавляющее большинство пациентов, оказавшихся в подобной ситуации, предпочли бы следовать рекомендуемым путем, и лишь небольшая часть из них отвергли бы этот путь
Подавляющему большинству своих пациентов врач будет рекомендовать следовать именно этим путем
Рекомендация может быть принята в качестве стандарта действия медицинского персонала в большинстве клинических ситуаций
Уровень 2
«Эксперты полагают»
Большая часть пациентов, оказавшихся в подобной ситуации, высказались бы за то, чтобы следовать рекомендуемым путем, однако значительная часть отвергла бы этот путь
Для разных пациентов следует подбирать различные варианты рекомендаций, подходящие именно им. Каждому пациенту необходима помощь в выборе и принятии решения, которое будет соответствовать ценностям и предпочтениям данного пациента
Рекомендации, вероятно, потребуют обсуждения с участием всех заинтересованных сторон до принятия их в качестве клинического стандарта
«Нет градации»
(НГ)
Данный уровень применяется в тех случаях, когда в основу рекомендации укладывается здравый смысл исследователя-эксперта или тогда, когда обсуждаемая тема не допускает адекватного применения системы доказательств, используемых в клинической практике.
Таблица 6. Оценка качества доказательной базы (составлена в соответствии с клиническими рекомендациями KDIGO).
Качество доказательной базы
Значение
А – высокое
Эксперты уверены, что ожидаемый эффект близок к рассчитываемому
В — среднее
Эксперты полагают, что ожидаемый эффект близок к рассчитываемому эффекту, но может и существенно отличаться
С – низкое
Ожидаемый эффект может существенно отличаться от рассчитываемого эффекта
D – Очень низкое
Ожидаемый эффект очень неопределенный и может быть весьма далек от рассчитываемого
Приложение Б. Алгоритмы ведения пациента
Алгоритм ведения больных с БМИ (дети)
«Диагностика»
н
нет да
нет да
Нет Да
«Лечение»
нет да
нет да нет да
Приложение В. Информация для пациентов
- Рекомендация II1. Для своевременной диагностики рецидива заболевания с целью мониторирования протеинурии рекомендовано использовать в домашних условиях определения белка с помощью тест-полосок.
- Рекомендация II2. На фоне заболевания ОРВИ доза глюкокортикостероидов снижать не следует, а при стероидзависимой форме возможен переход на ежедневный прием в той же дозе коротким курсом (на период заболевания),с последующим переходом на альтернирующий прием.
- Рекомендация II3. При обострении заболевания не следует самостоятельно подбирать дозу иммуносупрессивных препаратов. Коррекция лечения должна проводиться либо в специализированном стационаре, либо в амбулаторных условиях врачом-специалистом.
- Рекомендация II4. Отдых проводить в климатических условиях близких к климату проживания.
Приложение Г.
8.2. Гломерулонефриты
Гломерулонефриты (ГН) — неоднородная группа приобретенных заболеваний почек, различных по этиологии, клинико-морфологическим проявлениям, течению и исходу, преимущественно носящих характер иммунного воспаления с первичным поражением клубочков и вторичным вовлечением в патологический процесс канальцев и интерстиция.
Эти заболевания — одна из ведущих причин формирования хронической болезни почек, под которой понимают наличие любых клинических и лабораторных маркеров повреждения почек, персистирующих более 3 мес, вне зависимости от нозологического диагноза.
Классификация
ГН может быть:
-
первичным (идиопатическими), т.е. самостоятельным заболеванием;
-
вторичным, развившимся в рамках системных (СКВ, пурпура Шенляйна–Геноха и др.) или иных заболеваний (хронический ВГВ, БК, анкилозирующий спондилоартрит и др.).
По течению выделяют:
-
острый;
-
быстропрогрессирующий;
-
хронический ГН.
Ведущей является морфологическая классификация, основанная на результатах исследования почечного биоптата. ГН, при которых выявляют морфологические признаки воспаления, определяемые по наличию гиперклеточности клубочка, обусловленной пролиферацией собственных (резидентных) клеток и лейкоцитарной инфильтрацией (нейтрофилами, моноцитами, реже лимфоцитами), считают пролиферативными. Они могут быть эндокапиллярными, когда увеличено количество эндотелиальных и мезангиальных клеток, и экстракапиллярными, когда увеличено число париетальных эпителиальных клеток. Эти заболевания чаще всего проявляются нефритическим синдромом. К ним относят:
-
диффузный пролиферативный;
-
мезангиопролиферативный;
-
экстракапиллярный;
-
мембранопролиферативный (занимает промежуточное положение, так как сочетает пролиферацию клеток клубочка и поражение базальных мембран).
Для непролиферативных ГН характерно поражение слоев клубочкового фильтра (подоцитов и базальной мембраны), служащих основным барьером для белков. К ним относят:
-
болезнь минимальных изменений;
-
фокально-сегментарный гломерулосклероз;
-
мембранозную нефропатию.
Эти заболевания проявляются нефротическим синдромом.
Однако однозначной связи морфологических вариантов с этиологией, клинической картиной и патогенезом нет. Многие из них имеют несколько этиологических факторов, иммунных механизмов и клинических проявлений.
Этиология
Этиология большинства форм ГН неизвестна.
Ее чаще можно установить при остром процессе (в 80–90% случаев) и очень редко (в 5–10%) при хроническом. Среди этиологических факторов имеют значение стрептококковая, стафилококковая и другие бактериальные инфекции. Доказана роль вируса ВГВ и HCV, вирусов герпеса (1-го и 2-го типов, цитомегаловируса), ВИЧ, энтеровирусов. Возможно развитие ГН на фоне паразитарных заболеваний (например, при малярия); токсического воздействия лекарств (препаратов золота, пеницилламина, НПВП и др.), у подростков — алкоголя, наркотиков. Иногда начало заболевания бывает спровоцировано неинфекционными факторами (профилактическими прививками, введением сыворотки, охлаждением и т.д.), вызывающими аллергическую реакцию или служащими «пусковым» моментом на фоне предшествующей сенсибилизации организма.
Патогенез
Инфекционные или иные стимулы вызывают иммунный ответ, представленный иммунокомплексным или антительным механизмом.
-
Иммунокомплексный механизм заключается в образовании иммунных комплексов, состоящих из Аг и синтезированных против них АТ, которые могут формироваться в крови больного (циркулирующие иммунные комплексы) и затем фиксироваться на базальной мембране клубочков и в мезангии или непосредственно в гломерулярной ткани (in situ). Образованные иммунные комплексы классическим или альтернативным путем активируют систему комплемента, вследствие чего происходит выброс факторов, обеспечивающих хемотаксис и адгезию полиморфноядерных лейкоцитов и тромбоцитов, дегрануляцию базофилов и тучных клеток, формирующих мембраноатакующий комплекс (С5b–C9), непосредственно повреждающий гломерулярную базальную мембрану. Это приводит к активации тромбоцитов, свертывающей и калликреин-кининовой систем крови.
-
Антительный механизм обусловлен фиксацией цитотоксических АТ, направленных против собственных Аг в тканях клубочка с последующей активацией комплемента и распространением иммуновоспалительного повреждения. «Классический» Аг при антительном ГН — гликопротеин ГБМ. Наряду с этим цитотоксические АТ могут связываться с Аг подоцитов и мезангиальных клеток.
-
В клубочках накапливается множество клеток (нейтрофилов, эозинофилов, моноцитов-макрофагов, тромбоцитов), продуцирующих в большом количество различные медиаторы воспаления: цитокины (ФНОα, ИЛ-1, ИЛ-6, интерферон γ), факторы роста (тромбоцитарный фактор роста, трансформирующий фактор роста β), протеолитические ферменты, активные радикалы кислорода, липидные медиаторные субстанции, провоспалительные простагландины, вазоактивные субстанции. Цитокины и факторы роста вырабатываются как инфильтрирующими воспалительными клетками так и собственными клетками клубочков и интерстиция.
-
Одновременно с пролиферацией клеток клубочков (мезангиальных, эндотелиальных или эпителиальных) усиливается синтез (экспансия) внеклеточного матрикса. При длительном воспалении развиваются гломерулосклероз и интерстициальный фиброз — морфологическая основа хронической почечной недостаточности.
-
В прогрессировании почечного повреждения важную роль играют неиммунные механизмы:
-
гемодинамические факторы — внутриклубочковая гипертензия и гиперфильтрация, связанные с повышением системного АД и гиперфункцией оставшихся нефронов; они усиливают проницаемость гломерулярного фильтра, что способствует отложению различных макромолекул плазмы в тканях нефрона, ведут к активации ренин-ангиотензин-альдостероновой системы, повышению синтеза ангиотензина II — важного фактора пролиферации клеток почечных клубочков;
-
выраженная, длительная протеинурия действует как «внутренний токсин», поскольку реабсорбция профильтровавших белков активирует эпителий проксимальных канальцев, что стимулирует высвобождение им воспалительных и вазоактивных веществ — хемокинов и эндотелина; последние, синтезируясь в большом количестве, привлекают другие клетки, вызывающие воспалительную интерстициальную реакцию, предшествующую развитию тубулоинтерстициального фиброза;
-
гиперлипидемия оказывает повреждающее действие на эндотелий капилляров клубочков, а продукты перекисного окисления липидов стимулируют пролиферацию мезангия и синтез коллагена (развитие гломерулосклероза).
-
Воздействие на патогенетические иммунные процессы и неиммунные факторы прогрессирования изменений в почках — основной принцип лечения ГН, заключающийся:
-
в иммуносупрессии (глюкокортикоиды, цитостатики);
-
нефропротекции (ингибиторы ангиотензин-превращающего фермента, блокаторы рецепторов ангиотензина, обладающие гипотензивным, антипротеинурическим и антисклеротическим эффектом).
8.2.1. Острый постстрептококковый гломерулонефрит
Самый частый вариант ГН в детском возрасте. Ежегодно в мире выявляют 470 тыс. новых случаев острого постстрептококкового ГН, из них 400 тыс. — у детей. Наиболее высокая заболеваемость в возрасте от 5 до 12 лет, мальчики болеют чаще девочек. Обычно наблюдают спорадические случаи заболевания, но возможны и эпидемические вспышки.
Этиология
Этиология — БГСА.
В основном нефритогенные М-штаммы: 1, 2, 4, 12, 49, 55 57, 60. Это подтверждают бактериологическое исследование (выделение чистой культуры стрептококка из зева, из очагов пиодермии) и высокие уровни АТ к стрептококку (антистрептолизина О, анти-ДНКазы В, АТ к М-протеину и др.). Предполагают, что нефротропными Аг являются нефрит-ассоциированный рецептор плазминового комплекса, стрептококковый пирогенный экзотоксин В и его предшественник зимоген. Возможно развитие острого постстрептококкового ГН, вызванного β-гемолитическим стрептококком группы С.
Заболевание развивается через 1–3 нед после фарингеальной (ангина, фарингит, скарлатина) или через 3–6 нед после кожной инфекции (пиодермии). Возможно развитие острого постстрептококкового ГН после отита, лимфаденита, остеомиелита.
Патогенез
Патогенез: иммунокомплексное заболевание, механизмом развития которого может быть:
-
первичная фиксация Аг нефритогенных штаммов стрептококков в клубочках почек (в гломерулярной базальной мембране и/или мезангиуме), связывание их с аутоантителами с образованием иммунных комплексов in situ и активацией комплемента (данный иммунный процесс наиболее вероятен);
-
отложение в гломерулах циркулирующих иммунных комплексов, в состав которых входит стрептококковый Аг;
-
фиксация стрептококкового Аг в почечной ткани с развитием феномена молекулярной мимикрии с перекрестным взаимодействием АТ с гломерулярными структурами.
Морфология
Морфология: диффузный пролиферативный эндокапиллярный ГН с лейкоцитарной (преимущественно нейтрофильной) инфильтрацией капиллярных петель (при световой микроскопии, рис. 8.4).
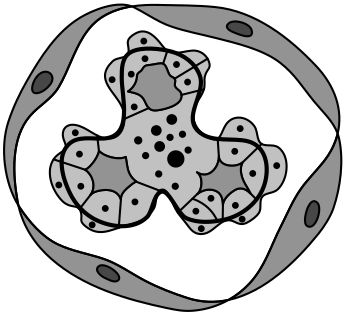
Рис. 8.4. Схема морфологии острого постстрептококкового гломерулонефрита (К.А. О’Каллагхан, 2009)
При иммунофлюоресцентном исследовании обнаруживают диффузное отложение IgG и C3-компонента комплемента гранулярного характера в мезангии и капиллярной стенке. Патогномоничный признак острого постстрептококкового ГН при электронной микроскопии — субэпителиальные плотные куполообразные депозиты в виде горбов. Возможно (в редких случаях) формирование полулуний, определяющее неблагоприятный прогноз заболевания.
Клиническая картина
Клиника: остронефритический синдром или бессимптомная гематурия.
Заболевание дебютирует внезапно после перенесенной стрептококковой инфекции. Возникают симптомы интоксикации (недомогание, вялость, плохой аппетит, тошнота, бледность, субфебрильное повышение температуры тела), могут быть боли в поясничной области, причина которых — растяжение капсулы почек вследствие отека их паренхимы.
При типичном циклическом течении острого постстрептококкового ГН развивается остронефритический синдром, который включает экстраренальные и ренальные симптомы.
Экстраренальные (клинические) симптомы:
-
отеки появляются у большинства пациентов, преимущественно на лице (более заметные по утрам), иногда — распространенные с развитием асцита и гидроторакса, что чаще наблюдается у детей младшего возраста; причина отеков — гиперволемия вследствие снижения фильтрации и задержки натрия;
-
макрогематурия (в 50% случаев) — моча становится темно-коричневой (что связано с изменением цвета гемоглобина в кислой среде), реже приобретает красный цвет и имеет вид «мясных помоев» (при щелочной реакции мочи);
-
АГ (у 75% пациентов) — повышено как систолическое, так и диастолическое АД. АГ обусловлена увеличением объема циркулирующей крови из-за задержки натрия и жидкости, а также повышением сердечного выброса и периферического сопротивления сосудов.
Ренальные (лабораторные) симптомы:
-
гематурия разной степени (от микро- до макро-), обязательный симптом у всех детей;
-
цилиндрурия (в основном эритроцитарные цилиндры);
-
протеинурия, обычно не превышающая 1 г в сут (субнефротического уровня);
-
олигурия (уменьшение объема выделяемой мочи), причины которой — снижение клубочковой фильтрации, задержка натрия и воды.
В начале заболевания возможна абактериальная лейкоцитурия (в основном лимфоцитурия), отражающая острый иммуновоспалительный процесс в клубочках. В редких случаях (у 2–4% пациентов) развивается нефротический синдром.
Лабораторная данные:
-
повышение уровня АТ к Аг стрептококка (антистрептолизина О, анти-ДНКазы В и др.); пик антистрептолизина О достигается через 2–4 нед после фарингита, уровень его остается повышенным в течение нескольких месяцев;
-
гипокомплементемия — снижение активности С3 и общей гемолитической активности комплемента наблюдают почти у всех в течение 4–8 нед;
-
умеренный лейкоцитоз (вторичный по отношению к перенесенной стрептококковой инфекции), иногда повышение СОЭ, снижение уровней гемоглобина и тромбоцитов в результате гемодилюции.
В типичных случаях острый постстрептококковый ГН имеет циклическое течение — вслед за остронефритическим периодом происходит обратное развитие симптомов, затем наступает клинико-лабораторная ремиссия. Отеки и АГ обычно купируются быстро (в течение 10–14 дней), изменения в моче (протеинурия, микрогематурия) исчезают через несколько месяцев (1,5–2, иногда позже).
В настоящее время заболевание чаще имеет ациклическое течение с изолированным мочевым синдромом (микрогематурия, цилиндрурия, протеинурия менее 1 г/сут). Экстраренальные проявления отсутствуют либо выражены столь незначительно и кратковременно, что остаются незамеченными.
При тяжелом течении острого ГН в начальном период возможны угрожающие жизни осложнения:
-
гипертензионная энцефалопатия — эклампсия, обусловленная спазмом сосудов головного мозга и его отеком (головная боль, рвота, снижение зрения, возможны тонико-клонические судороги, потеря сознания); при отсутствии своевременной адекватной терапии возможен летальный исход от кровоизлияния в головной мозг;
-
острое повреждение почек, проявляющееся резким сокращением или прекращением мочевыделения (анурия), снижением скорости клубочковой фильтрации, гипергидратацией, азотемией (повышением уровней мочевины и креатинина сыворотки), гиперкалиемией, метаболическим ацидозом;
-
острая сердечно-сосудистая недостаточность — у детей это осложнение возникает редко.
Биопсия почки целесообразна при долго сохраняющихся экстраренальных симптомах, изменениях в моче, сочетании нефритического синдрома с нефротическим, длительной депрессии С3, отсутствии восстановления скорости клубочковой фильтрации и уровня азотистых шлаков через 2–3 нед от дебюта болезни. Последнее требует исключения быстропрогрессирующего ГН, отсроченная терапия которого приводит к быстрому и необратимому нарушению почечных функций.
Острый постстрептококковый ГН следует дифференцировать от других пролиферативных вариантов ГН, проявляющихся остронефритическим синдромом (экстракапиллярного, мембранопролиферативного) или изолированной гематурией (IgA-нефропатии), наследственных гломерулопатий (болезнь тонких базальных мембран, синдром Альпорта), мочекаменной болезни.
Лечение
Лечение больных острым постстрептококковым ГН проводят в стационаре.
-
В остром периоде — постельный режим до купирования экстраренальных симптомов (исчезновения отеков, снижения АД).
-
Ограничение приема жидкости, натрия хлорида, белка. Объем жидкости рассчитывают по диурезу предыдущего дня, учитывая потери на перспирацию. В первые дни болезни при олигурии, АГ, распространенных отеках, снижении клубочковой фильтрации назначают бессолевой стол (после купирования этих симптомов соль постепенно добавляют — 0,5– 1 г/сут), ограничивают белок до 1,0–0,5 г/кг в сут, продукты, богатые калием (в том числе фруктовые и овощные соки). Общая энергетическая ценность пищи должна соответствовать потребностям ребенка в основном за счет углеводов и жиров.
-
Антибактериальная терапия показана при сохранении активности стрептококковой инфекции к моменту диагностики ГН, положительных результатах бактериологического исследования (мазка из зева или посева из очагов пиодермии) на БГСА. Используют препараты с низкой нефротоксичностью, дозу определяют с учетом скорости клубочковой фильтрации. Препараты первого выбора — антибиотики пенициллинового ряда (амоксициллин + клавулановая кислота, ампициллин и др.), курс 7–10 дней. Альтернативные препараты (второго выбора): макролиды 2-го и 3-го поколений (азитромицин, кларитромицин), цефалоспорины 2-го поколения (цефуроксим и др.).
-
Диуретическую терапию назначают при выраженных отеках, АГ: петлевые диуретики — фуросемид (1,5–2 мг/кг в сут парентерально 1–2 дня, затем внутрь 3–5 дней). При скорости клубочковой фильтрации выше 30 мл/мин можно использовать тиазидные препараты. Применение калийсберегающих диуретиков ограничено риском развития гиперкалиемии.
-
Гипотензивные средства необходимы при выраженной АГ, когда диуретической терапии недостаточно для контроля АД. Используют блокаторы медленных кальциевых каналов (нифедипин 0,25–0,5 мг/кг в сут, амлодипин и др.) или β-адреноблокаторы. Назначение ингибиторов ангиотензин-превращающего фермента и блокаторов ангиотензиновых рецепторов нежелательно из-за возможности развития гиперкалиемии, снижения скорости клубочковой фильтрации. При эклампсии для получения быстрого гипотензивного эффекта вводят прямые вазодилататоры (гидралазин и др.).
-
Возможно назначение антиагрегантов для улучшения почечного кровотока [дипиридамол, пентоксифиллин (Трентал♠)], антикоагулянтов — при выраженной гиперкоагуляции, связанной с развитием нефротического синдрома.
-
При остром повреждении почек, нарастающей азотемии, неконтролируемой гиперкалиемии необходимо проведение диализа.
Прогноз
Прогноз обычно хороший, большинство (85–90%) детей выздоравливает. У некоторых пациентов возникает быстропрогрессирующий (экстракапиллярный) ГН с развитием прогрессирующей хронической болезни почек.
8.2.2. Быстропрогрессирующий гломерулонефрит
Быстропрогрессирующий ГН (подострый, злокачественный, экстракапиллярный, с полулуниями) характеризуется чрезвычайно высокой активностью, тяжелым прогрессирующим течением, нарастающей почечной недостаточностью с развитием терминальной уремии в течение нескольких недель или месяцев.
Быстропрогрессирующий ГН у детей как вариант первичного ГН наблюдают редко (в 1–2% случаев), преимущественно у подростков; вторичный — может развиться при различных инфекционных, системных заболеваниях, опухолях. Чаще всего это конечная стадия острого постстрептококкового ГН и синдрома Гудпасчера.
Этиология и патогенез
Выделяют несколько иммунопатогенетических типов быстропрогрессирующего ГН в зависимости от наличия или отсутствия иммунных депозитов в клубочках почек, а также характера их свечения при иммунофлюоресцентной микроскопии:
-
с АТ против Аг базальной мембраны клубочков при идиопатическом быстропрогрессирующем ГН, синдроме Гудпасчера (АТ циркулируют в сыворотке крови и выявляются в биоптате почки — линейное свечение);
-
иммунокомплексный, наиболее типичный для острого постстрептококкового ГН, криоглобулинемии, СКВ, IgA-нефропатии, пурпуры Шенляйна–Геноха, мембранопролиферативного ГН; в мезангии и капиллярной стенке обнаруживают депозиты иммунных комплексов, имеющие гранулярное свечение;
-
без иммунных депозитов (малоиммунный), но часто с АТ к компонентам цитоплазмы нейтрофилов (протеиназе 3, миелопероксидазе), которые определяют в сыворотке крови, что характерно для микроскопического полиангиита, гранулематоза Вегенера.
Морфология
Патологоанатомическая особенность этого варианта — пролиферация клеток париетального эпителия (экстракапиллярный) с образованием более чем в 50% клубочков полулуний, служащих гистологическим маркером быстропрогрессирующего ГН (рис. 8.5).
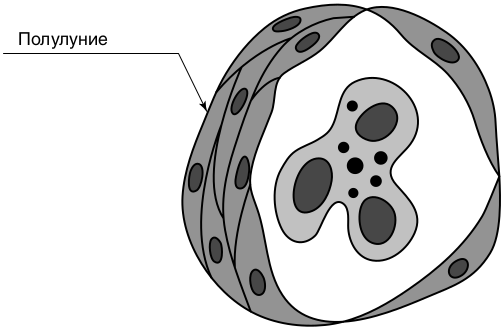
Рис. 8.5. Схема морфологии экстракапиллярного гломерулонефрита (О’Каллагхан К.А., 2009)
Полулуния — следствие тяжелого повреждения клубочков, вызванного АТ к базальной мембране или иммунными комплексами, или АТ к компонентам цитоплазмы нейтрофилов, приводящего к разрыву стенок капилляров. В результате в пространство капсулы Боумена попадают плазменные белки и воспалительные клетки (в основном пролиферирующие париетальные эпителиальные клетки и макрофаги), фибрин, формирующие инфильтрат с образованием дугообразных утолщений капсулы клубочка — полулуний (получивших такое название из-за характерного внешнего вида на срезах клубочков).
Последние окружают клубочковые капилляры и вызывают их спадение. При преобладании в полулуниях макрофагов, разрыве капсулы из интерстиция поступают фибробласты и миофибриллы, синтезирующие матриксные белки (коллаген типов 1 и 3, фибронектин), что ведет к необратимому фиброзу полулуний.
Клиническая картина и диагностика
Клинические и лабораторные признаки:
-
остронефритический синдром (см. острый постстрептококковый ГН);
-
быстропрогрессирующая почечная недостаточность, критерий которой — повышение уровня креатинина сыворотки в 2 раза за каждые 3 мес болезни или в более короткий срок.
АГ высокая, носит стойкий характер. Протеинурия неселективная, субнефротического уровня, иногда развивается нефротический синдром. Рано, с первых месяцев, а иногда и с первых недель болезни (2–3-й), появляются клинические и лабораторные симптомы почечной недостаточности:
-
слабость, утомляемость, снижение аппетита, анорексия, тошнота, рвота;
-
снижение скорости клубочковой фильтрации, азотемия, гиперкалиемия, нарушения водно-электролитного обмена, метаболический ацидоз, анемия, вторичный гиперпаратиреоз.
Терминальная уремия развивается в течение года от дебюта заболевания. Результаты иммунологического исследования крови зависят от иммунопатогенетического типа быстропрогрессирующего ГН: при иммунокомплексном отмечают снижение концентрации С3-компонента и гемолитической активности комплемента, при антительном — обнаруживают АТ к базальной мембране (к коллагену 4-го типа), при малоиммунном — АТ к компонентам цитоплазмы нейтрофилов.
Лечение
Лечение быстропрогрессирующего ГН следует начинать безотлагательно ввиду чрезвычайно высокой активности процесса и быстрой необратимой потери функции почек.
Показана интенсивная иммуносупрессивная терапия, которую начинают, не дожидаясь результатов иммунологии крови и нефробиопсии. С этой целью, согласно международному стандарту, используют:
-
пульс-терапию метилпреднизолоном (20 мг/кг в сутки, но не более 1000 мг на введение, внутривенно капельно 3 дня подряд) с последующим переходом на пероральный прием (1,0–0,5 мг/кг в сутки), курсы пульс-терапии повторяют через 3–4 недели.
После верификации диагноза лечение глюкокортикоидами сочетают с цитостатиками:
-
пульс-терапией циклофосфамидом (15 мг/кг в сутки, внутривенно 1 раз в 2 нед 3 раза, далее — в 3–4 нед) или препарат назначают перорально (2 мг/кг в сутки) до достижения стойкого улучшения.
Комбинированная иммуносупрессивная терапия рекомендуется при иммунокомплексном и малоиммунном патогенетических типах быстропрогрессирующего ГН.
-
Интенсивный плазмоферез в дополнение к глюкокортикоидам целесообразно использовать при быстропрогрессирующем ГН, обусловленном образованием АТ к базальной мембране клубочков почек.
При неэффективности лечения и развитии терминальной почечной недостаточности начинают программный гемодиализ и выполняют трансплантацию почки.
Дифференциальную диагностику быстропрогрессирующего ГН необходимо проводить с заболеваниями, приводящими к острому повреждению почек, такими как острый постстрептококковый ГН, острый интерстициальный нефрит, острый тубулярный некроз, атипичный гемолитико-уремический синдром и др.
Прогноз
Прогноз, даже при своевременно начатом активном лечении, остается тяжелым из-за высокого риска быстрого исхода в терминальную почечную недостаточность. Для его оценки необходимо проведение биопсии почек. При быстропрогрессирующем ГН, ассоциированном с острым постстрептококковым ГН, возможен благоприятный исход в ремиссию с восстановлением почечных функций.
8.2.3. Хронический гломерулонефрит
Хронический ГН — группа разнородных первичных гломерулопатий, характеризующихся прогрессирующими, деструктивными, склеротическими изменениями с постепенным ухудшением почечных функций и исходом в хроническую почечную недостаточность.
Пролиферативные гломерулонефриты
Мезангиопролиферативный гломерулонефрит (иммуноглобулин А-нефропатия)
IgA-нефропатия может быть самостоятельным заболеванием (первичная IgA-нефропатия — болезнь Берже) и вторичной — при многих системных (пурпура Шенляйна–Геноха, СКВ и др.) и хронических заболеваниях (ВГВ и гепатит С, ЯК, БК, МВ, саркоидоз и др.).
IgA-нефропатия (болезнь Берже) — самая распространенная форма первичного ГН.
Заболевание может дебютировать в любом возрасте. Мальчики болеют в 2 раза чаще девочек.
Морфология: пролиферация мезангиальных клеток (очаговая или диффузная), расширение мезангия, отложение иммунных комплексов в мезангии и под эндотелием (рис. 8.6). Чаще всего в клубочках выявляют депозиты IgА — IgA-нефропатия, с которой в настоящее время отождествляют данный морфологический вариант ГН.
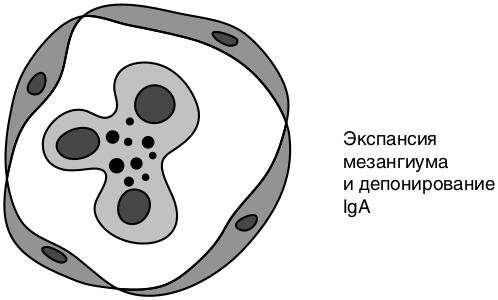
Рис. 8.6. Схема морфологии мезангиопролиферативного гломерулонефрита (О’Каллагхан К.А., 2009); Ig — иммуноглобулин
Механизм развития IgA-нефропатии — иммунокомплексный, в развитии которого ведущая роль принадлежит увеличению синтеза и изменению структуры молекулы IgA, вызванному нарушением процессов ее гликозилирования и полимеризации. IgA-полимеры и IgA-содержащие иммунные комплексы длительно находятся в циркуляции, не выводятся клетками ретикулоэндотелиальной системы, депонируются в мезангии, в результате чего активируется синтез клетками почек различных цитокинов, факторов роста, развиваются характерные морфологические изменения.
Клиническая картина IgA-нефропатия разнообразна. Наиболее частые клинические варианты следующие.
-
Макрогематурия — в виде повторных эпизодов (рецидивирующая), сопровождающих респираторную инфекцию (синфарингитная). Макрогематурия возникает одновременно или в первые дни болезни (2–3-й) и сохраняется от нескольких часов до нескольких дней. Моча обычно бурого цвета. Рецидивы могут провоцироваться вакцинацией, физической нагрузкой. Между эпизодами синфарингитной макрогематурии в анализах мочи выявляют микрогематурию.
-
Персистирующая микрогематурия, иногда в сочетании небольшой протеинурией и/или АГ.
При фазово-контрастной микроскопии осадка мочи обнаруживают дисморфные эритроциты, указывающие на гломерулярное происхождение гематурии. У 35–60% больных отмечают повышение уровня IgА в сыворотке крови. Тактика лечения IgA-нефропатия основана на оценке риска прогрессирования заболевания. При изолированной гематурии лечение не назначают. При гематурии, в том числе с эпизодами синфарингитной макрогематурии, небольшой протеинурии (0,5–1 г/сут), нормальной скорости клубочковой фильтрации и отсутствии АГ целесообразна нефропротективная терапия. При протеинурии нефротического уровня показана иммуносупрессивная терапия — глюкокортикоиды в течение 6 мес. Необходима санация очагов инфекции, провоцирующих обострения заболевания.
У детей прогноз IgA-нефропатии (болезни Берже) обычно благоприятный, особенно если она проявляется синфарингитной гематурией.
Дифференциальную диагностику прежде всего следует проводить с наследственными гломерулопатиями — болезнью тонких базальных мембран и синдромом Альпорта. Болезнь тонких базальных мембран (семейная доброкачественная гематурия) — заболевание с аутосомно-доминантным типом наследования, проявляющееся изолированной микрогематурией и имеющее благоприятное течение. Его диагностируют по фокальному или диффузному истончению гломерулярных базальных мембран, которое выявляют при электронной микроскопии нефробиоптатов, иммунный материал в ткани почек отсутствует. Синдром Альпорта (Х-сцепленный, аутосомно-рецессивный и аутосомно-доминантный варианты) — генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген 4-го типа базальных мембран. Проявляется гематурией и/или протеинурией, имеет прогрессирующее течение с исходом в почечную недостаточность. Поражение почек может сочетаться с двусторонней нейросенсорной тугоухостью и патологией зрения (передний лентиконус, крапинки на желтом пятне и эрозии роговицы).
Мембранопролиферативный гломерулонефрит
Мембранопролиферативный (мезангиокапиллярный) ГН — иммунокомплексное заболевание, морфологические особенности которого — мезангиальная пролиферация и утолщение стенок капилляров с образованием двухконтурных базальных мембран за счет проникновения в них мезангиальных клеток (рис. 8.7). При электронной микроскопии выявляют субэндотелиальные депозиты иммунных комплексов (1-й тип) или плотные отложения иммунных комплексов внутри базальной мембраны клубочков (2-й тип, болезнь плотных депозитов).
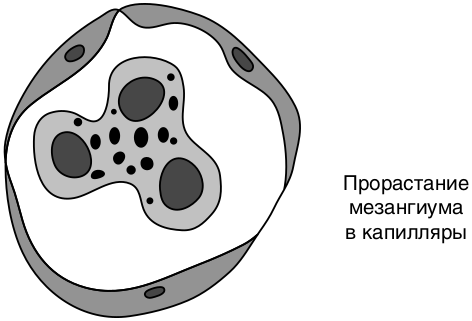
Рис. 8.7. Схема морфологии мембранопролиферативного гломерулонефрита (О’Каллагхан К.А., 2009)
Прогрессирование патологического процесса приводит к развитию склероза и формированию фибропластического ГН — финалу большинства форм хронического ГН. Развивается склероз капиллярных петель клубочка, формируются фиброэпителиальные и фиброзные полулуния, утолщение и склероз капсулы клубочка.
Обычно мембранопролиферативный ГН диагностируют у детей 10–12 лет. Нередко устанавливают связь мембранопролиферативного ГН (1-й тип) с инфицированием вирусом ВГВ и HCV. Этот тип ГН выявляют при СКВ, синдроме Шегрена, саркоидозе, лимфоме.
Особенности морфологии (сочетание пролиферация мезангия с поражением базальных мембран клубочков) определяют клиническую картину мембранопролиферативного ГН — сочетание нефритического и нефротического синдромов. Для первичного и вторичном вариантов заболевания характерны: стойкая, высокая АГ, выраженная неселективная протеинурия, гипокомплементемия со снижением уровня С3- и/или С4-компонентов комплемента, анемия, криоглобулинемия (особенно у больных гепатитом С).
При выборе терапии идиопатического мембранопролиферативного ГН учитывают клиническое течение заболевания и результаты биопсии почки. При ведущем нефротическом синдроме (НС), медленном снижении функции почек рекомендуют циклофосфамид (2–2,5 мг/кг в сут) или микофенолата мофетил в сочетании с преднизолоном в альтернирующем режиме. При нефритическом синдроме с быстропрогрессирующим падением почечных функций вначале проводят плазмаферез, пульс-терапию метилпреднизолоном (3 дня), далее продолжают иммуносупрессивное лечение по предыдущей схеме.
Мембранопролиферативный ГН — один из самых неблагоприятных вариантов ГН с быстрым исходом (у 50% больных) в терминальную почечную недостаточность.
Непролиферативные гломерулонефриты
Непролиферативные ГН обусловлены иммунопатологическими процессами, но при этом нет морфологических признаков воспаления, поскольку пролиферации собственных клеток клубочка отсутствует или выражена минимально. Поэтому в ряде классификаций их относят к группе непролиферативных гломерулопатий.
Подоцитопатии — болезнь минимальных изменений и фокально-сегментарный гломерулосклероз (особенно в дебюте заболевания) — имеют сходную морфологическую картину, что позволило объединить их термином идиопатический нефротический синдром.
Нефротический синдром — симптомокомплекс, включающий высокую протеинурию (более 50 мг/кг в сут), гипоальбуминемию (ниже 25 г/л), гиперлипидемию и отеки. Распространенность его составляет 1 случай на 6000 детей, у мальчиков в 2 раза чаще, чем у девочек.
Болезнь минимальных изменений
Болезнь минимальных изменений имеет ряд синонимов: идиопатический нефротический синдром, ГН с минимальными изменениями, НС с минимальными изменениями, стероид-чувствительный НС, исторический термин — липоидный нефроз.
Болезнь минимальных изменений наблюдают в 80–90% случаев НС у детей младшей возрастной группы (до 10 лет), в 50% — у подростков. В структуре всех морфологических вариантов ГН у детей составляет 76,6%.
Морфология. Изменения клубочков выявляют только на ультраструктурном уровне (электронная микроскопия) в виде диффузного слияния (сглаживания) ножковых отростков подоцитов (подоцитопатия) и их микровиллезной трансформации (появления на эпителиальной поверхности многочисленных ворсинчатых образований, направленных в мочевое пространство); при световой микроскопии клубочек выглядит неизмененным, при флюоресцентном исследовании отсутствуют отложения Ig и фракций комплемента в структурах нефрона (рис. 8.8).
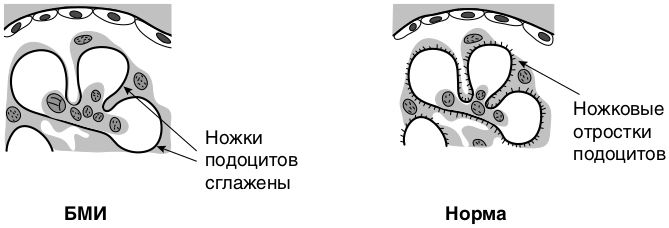
Рис. 8.8. Схема морфологии болезни минимальных изменений (О’Каллагхан К.А., 2009)
Болезнь минимальных изменений чаще всего бывает идиопатической.
Возможны вторичные формы, развивающиеся на фоне установленных причин: инфекций, лекарственных (чаще всего НПВП), токсических воздействий, атопии, злокачественных заболевания и др.).
Патогенетические механизмы идиопатической болезни минимальных изменений:
-
дисфункция Т-клеточного звена иммунитета, приводящая к продукции «фактора проницаемости», вызывающего дезорганизацию цитоскелета подоцитов, следствием чего становится увеличение диффузии альбумина через гломерулярную базальную мембрану (ГБМ); механизмы активации клеточного иммунитета и сам «фактор проницаемости» остаются неизвестными;
-
генетически детерминированные изменения протеинов щелевой диафрагмы и актинового цитоскелета подоцитов, вследствие чего повышена проницаемость гломерулярного фильтра.
Клинические и лабораторные симптомы идиопатической болезни минимальных изменений следующие.
-
Внезапное появление НС без видимой причины. Иногда провоцирующими факторами могут быть атопические реакции, ОРИ.
-
Генерализованные отеки (рис. 8.9), часто прогрессирующие до анасарки с развитием асцита, гидроторакса, гидроперикарда. Кожа бледная, холодная на ощупь. Диурез снижен.
-
Протеинурия массивная (может достигать 20–30 г в сут и более), высокоселективная (альбуминурия).
-
Резкая гипоальбуминемия (не всегда пропорциональная выраженности протеинурии), определяющая тяжесть НС, и гиповолемия.
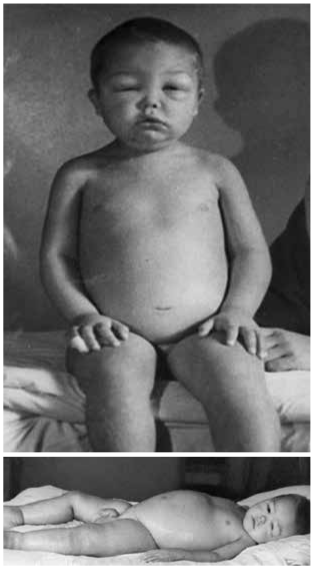
Рис. 8.9. Генерализованные отеки
Потеря белка (альбумина) с мочой приводит к снижению онкотического давления плазмы крови. Возникает онкотический градиент между внутрисосудистой и экстравазальной средой, вследствие чего происходит перемещение жидкости из сосудистого русла в ткани. Для восстановления объема циркулирующей крови запускаются компенсаторные механизмы (секретируется антидиуретический гормон, ингибируется синтез предсердного натрийуретического фактора, активируются ренин-ангиотензин-альдостероновая система (РААС), симпатическая нервная система), стимулирующие задержку натрия и воды в почках. Гипоальбуминемия усиливается (гипоальбуминемия разведения) и поддерживает отеки.
-
Выраженная гиперлипидемия с дислипопротеинемией (повышение концентрации общего холестерина, триглицеридов, липопротеидов низкой плотности, липопротеина А и снижение концентрации липопротеинов высокой плотности), причинами которых считают повышенный синтез липопротеидов низкой плотности, вызванный снижением онкотического давления, потерю с мочой липорегуляторных субстанций и снижение катаболизма липидов.
-
Симптомокомплекс НС не сопровождается ни АГ, ни гематурией («чистый» НС). Очень редко отмечают кратковременное повышение АД, связанное с задержкой натрия в ответ на гиповолемию, и микрогематурию.
-
Характерные лабораторные показатели:
-
резкое ускорение СОЭ (до 50–70 мм/ч);
-
уровни азотистых шлаков, С3-фракции комплемента нормальные.
-
Возможно развитие угрожающих жизни осложнений.
-
Гиповолемический шок, связанный с резким значительным падением объема циркулирующей крови: боли в животе, рвота, кожная эритема, гипотония, тахикардия, холодные конечности, уменьшение количества мочи.
-
Острое повреждение почек (см. острый постстрептококковый ГН), главная причина которого — гипоперфузия почек из-за гиповолемии.
-
Тромбозы венозные (почечных вен, глубоких вен нижних конечностей и др.), наблюдаемые чаще, и артериальные. Предполагают, что развитие их связано с гемоконцентрацией, повышением вязкости крови на фоне гиповолемии, гиперфибриногенемией, повышением агрегации тромбоцитов, потерей с мочой плазминогена, антитромбина III, протеинов С и S, угнетением фибринолиза. Способствуют их возникновению обездвиженность, лечение глюкокортикоидами и диуретиками.
-
Тяжелые инфекции вследствие вторичного иммунодефицита, вызванного потерей Ig и компонентов системы комплемента с мочой, дисфункцией Т-клеточного звена иммунитета, общих метаболических нарушений, применения иммуносупрессорных препаратов.
Наряду с этим развивается белково-энергетическая недостаточность, главная причина которой — гипоальбуминемия.
Течение болезни может быть острым с исходом в стойкую ремиссию и рецидивирующим.
Лечение. В активном периоде заболевания показан постельный режим. Объем жидкости дозируют в зависимости от диуреза. Назначают гипохлоридную диету или бессолевой стол. При резкой гиперлипидемии необходимо сократить прием жиров.
Диуретические препараты: петлевые диуретики (фуросемид, торасемид и др.).
Стандартное лечение первого эпизода болезни минимальных изменений [клинические рекомендации KDIGO (Kidney Disease: Improving Global Outcomes, научного общества нефрологов России)] — глюкокортикоидная терапия по схеме:
-
преднизолон в дозе 60 мг/м2 поверхности тела или 2 мг/кг (максимально 60 мг/сут) внутрь ежедневно в течение 4–6 нед; к этому времени у большинства детей наступает полная или частичная ремиссия (стероид-чувствительный НС);
-
снижение суточной дозы до 40 мг/м2 с переходом на альтернирующий режим (прием препарата через день), который используют в течение 4–6 нед;
-
продолжение постепенного снижения суточной дозы препарата до полной отмены.
Общая длительность терапии глюкокортикоидами должна составлять 4–5 мес.
Стероид-чувствительность при болезни минимальных изменений у детей составляет более 90%.
Поэтому для назначения инициальной терапии болезни минимальных изменений нет необходимости в предварительном проведении биопсии почки. Она возникает при подозрении на иные морфологические изменения в почечной ткани или вторичный характер болезни минимальных изменений.
Рецидив, развившийся после отмены глюкокортикоидов, свидетельствует о стероид-зависимости. Лечение первого рецидива НС проводят так же, как терапию в дебюте болезни. Отсутствие эффекта от терапии глюкокортикоидами в течение 8 нед указывает на резистентность НС к глюкокортикоидам. При частых рецидивах НС, стероид-зависимости и стероид-резистентности применяют стероидсберегающие препараты — алкилирующие агенты (циклофосфамид или хлорамбуцил), сохраняя прием самых низких доз преднизолона, необходимых для поддержания ремиссии.
При отсутствии терапевтического эффекта используют левамизол, ингибиторы кальцинейрина (циклоспорин или такролимус), микофенолата мофетил, ритуксимаб. При стероид-резистентном НС возможно проведение пульс-терапии метилпреднизолоном или комбинированной терапии различными иммуносупрессантами по индивидуальным схемам. Показано длительное применение ренопротективных препаратов [ингибиторов ангиотензин-превращающего фермента (фозиноприла, эналаприла)] и блокаторов ангиотензиновых рецепторов.
При угрозе тромботических осложнений (выраженный НС с гиповолемией, гиперфибриногенемией и гиперлипидемией) проводят профилактическое лечение антикоагулянтами: гепарины [далтепарин натрия (Фрагмин♠), надропарин кальция (Фраксипарин♠)], варфарин.
Показания к биопсии почки у детей с НС:
-
возраст ребенка до 1 года, когда велика вероятность генетических вариантов НС;
-
стероид-резистентность, которую констатируют при отсутствии эффекта от терапии глюкокортикоидами в дебютной дозе в течение 8 нед;
-
стероид-зависимость, свидетельство которой — рецидив НС при снижении дозы преднизолона или в течение 2 нед после его отмены;
-
сочетание НС с нефритическими симптомами;
-
прогрессирующее снижение почечных функций.
Прогноз при наличии чувствительности к глюкокортикоидам благоприятный — у большинства детей развивается стойкая ремиссия. Неблагоприятными факторами следует считать генетически обусловленную болезнь минимальных изменений, а также первичную и вторичную (отсутствие эффекта от глюкокортикоидов при рецидивах НС) стероид-резистентность, которые могут быть связаны с наличием фокально-сегментарного гломерулосклероза или трансформацией минимальных изменений в данную патологию.
Фокально-сегментарный гломерулосклероз
Фокально-сегментарный гломерулосклероз — главная причина (более 50% случаев) резистентности к глюкокортикоидам у детей.
Морфология: при световой микроскопии в начале заболевания изменения трактуют как минимальные (подоцитопатия), в последующем выявляют зоны склероза и гиалиноза в некоторых сегментах (сегментарные) отдельных (фокальные) клубочков (рис. 8.10).
Рис. 8.10. Схема морфологии фокально-сегментарного гломерулосклероза (О’Каллагхан К.А., 2009)
Сходство морфологии фокально-сегментарного гломерулосклероза и болезни минимальных изменений позволило предположить, что это разные стадии или разной тяжести варианты одного заболевания.
В развитии первичного варианта фокально-сегментарного гломерулосклероза ведущая роль принадлежит генетическому дефекту или действию циркулирующих факторов проницаемости (кардиотропиноподобному цитокину 1 из семейства ИЛ-6, растворимому рецептору к урокиназе и др.). Подоциты, подвергшиеся трансдифференциации, приобретают свойства макрофагов и участвуют в формировании очагов фиброза (скероза).
Клинико-лабораторные особенности фокально-сегментарного гломерулосклероза — НС или персистирующая субнефротическая протеинурия в сочетании с микрогематурией и АГ.
Основу терапии идиопатического фокально-сегментарного гломерулосклероза с НС составляют глюкокортикоиды (см. лечение болезни минимальных изменений). Эффективность терапии повышает применение высокодозных внутривенных пульсов метилпреднизолона.
Фокально-сегментарный гломерулосклероз — основная форма гломерулопатии, при которой развивается терминальная почечная недостаточность, требующая заместительной почечной терапии (программного диализа).
Прогноз неблагоприятный, особенно у детей с массивной протеинурией (более 10 г/сут). Кроме того, фокально-сегментарный гломерулосклероз очень часто (30–50%) рецидивирует в трансплантированной почке.
Мембранозная нефропатия
Мембранозная нефропатия в структуре НС у детей составляет менее 1%. Чаще развивается у детей школьного возраста (6–15 лет). Для мембранозной нефропатии характерно диффузное утолщение гломерулярной базальной мембраны (рис. 8.11) и изменение ее структуры из-за отложения иммунных депозитов под подоцитами (субэпителиально) и внутри мембраны (интрамембранозно).
Рис. 8.11. Схема морфологии мембранозной нефропатии (О’Каллаган К.А., 2009)
Механизм развития мембранозной нефропатии иммунокомплексный. Основа формирования иммунных комплексов при первичном форме патологии — образование аутоантител к внутренним Аг клубочка — подоцитарному трансмембранному рецептору фосфолипазы. Основные клинические проявления — НС или субнефротическая протеинурия.
При мембранозной нефропатии самый высокий риск тромботических осложнений НС. Возможны микрогематурия, АГ.
У детей заболевание чаще протекает без НС с нормальной функцией почек, часто приводит к развитию полной спонтанной ремиссии и не требует иммуносупрессивной терапии. Предикторы неблагоприятного прогноза — НС с массивной протеинурией, АГ и снижение почечных функций.
Тестовые задания
-
Для лечения первого эпизода нефротического синдрома при болезни минимальных изменений целесообразно назначить:
-
преднизолон 40 мг/м2 в сут;
-
преднизолон 60 мг/м2 в сут;
-
пульс-терапию метилпреднизолоном;
-
циклофосфамид.
-
-
Самая частая причина нефротического синдрома у детей:
-
болезнь минимальных изменений;
-
острый постстрептококковый ГН;
-
быстропрогрессирующий ГН;
-
мембранозная нефропатия.
-
-
Обязательное проявление остронефритического синдрома:
-
гематурия;
-
протеинурия;
-
лейкоцитурия;
-
АГ.
-
-
Остронефритический синдром характерен для:
-
болезни тонких базальных мембран;
-
острого пиелонефрита;
-
болезни минимальных изменений;
-
острого постстрептококкового ГН.
-
Ответы: 1 — b; 2 — a; 3 — a; 4 — d.
Литература
-
Детская нефрология : практическое руководство / под ред. Э. Лойманна, А.Н. Цыгина, А.А. Саркисяна. М. : Литтерра, 2010.
-
Нефрология. Клинические рекомендации / под ред. Е.М. Шилова, А.В. Смирнова, Н.Л. Козловской. М. : ГЭОТАР-Медиа, 2016.
-
О’Каллагхан К.А. Наглядная нефрология. М. : ГЭОТАР-Медиа, 2009.
-
Эрман М.В. Нефрология детского возраста : руководство для врачей. СПб. : СпецЛит, 2010.
Also known as lipoid nephrosis, nil disease, and minimal change nephropathy, the kidney histology on light microscopy in MCD is relatively normal and lacks the significant glomerular cell proliferation, infiltration by circulating immunoeffector cells, immune deposits, tubulointerstitial changes, or alterations in the glomerular basement membrane (GBM) that characterize other glomerular diseases.
From: National Kidney Foundation Primer on Kidney Diseases (Sixth Edition), 2014
Glomerular Diseases
A. Vishnu Moorthy, … Weixiong Zhong, in Pathophysiology of Kidney Disease and Hypertension, 2009
1 Minimal Change Nephrotic Syndrome
Previously thought to be lipid nephrosis caused by the cellular accumulation of lipids in the renal tubular cells, this condition tends to affect children and young adults. The patient with the minimal change nephrotic syndrome has proteinuria, edema, hypoalbuminemia, and hyperlipidemia. Hematuria, hypertension, and renal failure are uncommon.
Both light microscopy (Figs. 8-1 and 8-2) and immunofluorescence reveal no significant pathologic changes (hence the term minimal change). By contrast, the electron microscope shows effacement of the foot processes of the epithelial podocytes (Fig. 8-3). Minimal change disease is usually very responsive to therapy with antiinflammatory medications (e.g., prednisone). Although the microscopic results are illustrative of the condition, the presentation is well recognized now, so it is uncommon to perform diagnostic biopsies in children with the nephrotic syndrome unless the children’s conditions are refractory to treatment with prednisone.
Despite its well-characterized histology and ease of treatment, the pathogenesis of minimal change nephrotic syndrome is not clearly understood. Although circulating toxins have been suspected, the nature of these factors is unclear. What is known is that the pathology is a result of the loss of the net negative charge associated with the foot processes, thereby breaking down the glomerular capillary barrier.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B978141604391150014X
Non-Neoplastic Kidney
SHARDA G. SABNIS, … ZDENA PAVLOVA, in Modern Surgical Pathology (Second Edition), 2009
Minimal Change Disease (Lipoid Nephrosis, Visceral Epithelial Cell Disease, Foot Process Disease, Nil Disease, Minimal Change Glomerulopathy)
Disease Definition
Munk3 introduced the term lipoid nephrosis to describe a group of patients with heavy proteinuria but without glomerular changes by light microscopy. Perhaps the most prominent finding by light microscopy was the presence of lipid droplets in proximal tubular epithelium; hence the disease was called lipoid nephrosis. Because of the normal appearance of glomeruli or minimal mesangial cellularity, the term minimal change disease is used commonly.
Epidemiology
Minimal change disease accounts for approximately 80% of cases of nephrotic syndrome in children and 20% of cases in adults.4 In children, the average age is 3 years, and a male preponderance is reported.5,6
Cause/Etiology
Although most cases of minimal change disease are idiopathic, minimal change disease can occur secondary to specific causes, such as the following: lymphoma, including Hodgkin’s disease7,8; drugs such as nonsteroidal anti-inflammatory drugs (NSAIDs)9; and human immunodeficiency virus (HIV) infection. Viral upper respiratory tract infection is a common antecedent feature, and a history of allergy, atopy, or recent immunization may be found.10,11
Various theories have been proposed for the pathogenesis of minimal changes (and the presumed related diseases mesangial proliferative disease and focal segmental glomerulosclerosis [FSGS]).12-15 The reason for the increased permeability of the glomerular capillary wall to macromolecules in these diseases is not apparent. Although some decrease of glomerular polyanion has been demonstrated, the cause for this loss is not known. An experimental model of minimal change disease was developed by administration of the aminoglycoside puromycin, an agent specifically toxic to the glomerular epithelial cells that synthesize the negatively charged sialoproteins and proteoglycans so abundant in the glomerular basement membrane (GBM). Thus, it is believed that injury to the glomerular visceral epithelial cells results in reduction of glomerular polyanions, which in turn leads to a loss of the anionic charge barrier considered necessary to preventing anionic intravascular proteins, particularly albumin, from leaking across the GBM. Another hypothesis of pathogenesis is based on the association of minimal change disease with allergies, abnormal T-cell function, and abnormal cellular immunity. This hypothesis suggests that a circulating lymphokine or vasculotoxic substance produced by an aberrant clone of lymphocytes causes the disease. The rapid recurrence of FSGS in transplant recipients indicates that the idiopathic nephrotic syndrome is probably not a disease inherent in the kidneys but is related to an as yet unknown circulating factor that increases glomerular permeability.
Finally, and of considerable potential interest, it appears that mutations in certain podocyte-specific genes, such as nephrin (NPHS1) (chromosome 19) or podocin (NPHS2) (chromosome 1), which deal with maintaining the integrity of the slit diaphragm, which is the junction between visceral epithelial cells, can produce congenital and familial forms of nephrotic syndrome.16
Clinical Presentation
Patients typically present with nephrotic-range proteinuria (>3.0 to 3.5 g/24 hours). The proteinuria is often selective (mainly albuminuria) and is usually associated with hypoalbuminemia, lipiduria, and hyperlipidemia. The disease typically responds to corticosteroids, although relapses can occur on cessation of treatment. Nonetheless, the prognosis is excellent.
Pathology
All glomeruli are similar and are either normocellular or show a slight increase in endocapillary cells (minimal changes) (Fig. 29-11). The glomerular capillary walls are unremarkable, and the capillary lumens are widely patent. Swelling of visceral epithelial cells and edema in the renal interstitium are common findings. Tubules may contain hyaline (protein reabsorption) droplets or lipid droplets.
By electron microscopy, one sees diffuse or extensive effacement of epithelial cell foot processes and formation of villi on the external surface of the epithelial cells (so-called villous transformation) (Fig. 29-12). The glomerular capillary basement membranes are usually of normal thickness, without electron-dense deposits. Electron microscopy is essential for the diagnosis of minimal change disease. Results of immunofluorescence microscopic examination are negative for immunoglobulins and complement components.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9781416039662000291
Glomerular Disease
S. Akilesh, in Pathobiology of Human Disease, 2014
Minimal Change Disease
Minimal change disease was formerly known as lipoid nephrosis because of fatty infiltration of the kidney parenchyma at late stages and nil disease because of the minimal histologic findings seen on biopsy. Minimal change disease is the most common etiology for nephrotic syndrome in children. Patients generally present with heavy, nephrotic-range proteinuria that is selective, meaning that albumin is preferentially lost into the urine across the glomerular filtration barrier. Albumin is an anionic protein and is normally repulsed by the glomerular filtration barrier. Patients with minimal change disease exhibit a loss of anionic charge of the glomerular filtration barrier that may explain the increased passage of albumin over similarly sized neutral dextran molecules. On biopsy, light microscopy reveals no significant changes in the glomeruli – the peripheral capillary loops remain widely patent and the mesangial areas are unremarkable or show mild expansion (Figure 1). Tubules may show prominent protein reabsorption droplets. With longstanding disease, there is accumulation of lipid containing foamy macrophages (‘foam cells’) in the interstitium and foamy transformation of tubular epithelial cells (Figure 2). These changes likely contributed to the disease’s original description as lipoid nephrosis. Immunofluorescence studies are usually negative. The key diagnostic finding is diffuse effacement of the podocyte foot processes, detectable only by electron microscopy (Figure 3). On the urinary side, podocytes may exhibit membrane projections termed ‘apical microvilli,’ a change that is referred to as microvillous transformation (Figure 4). There is often a condensation of cytoplasmic filaments at the base of the effaced foot processes. The podocyte cytoplasm may be filled with large fluid and lipid vacuoles. These findings are not specific to minimal change disease and can be seen in FSGS and other glomerular diseases with heavy proteinuria. Minimal change disease is a less frequent cause of the nephrotic syndrome in adults, the reasons for which are unclear.

Figure 1. A histologically unremarkable glomerulus from a patient with minimal change disease (Jones methenamine silver (JMS), 600 × original magnification). By electron microscopy, the foot processes of the glomerular epithelial cells showed diffuse effacement (see Figure 3).

Figure 2. Foamy transformation of tubular epithelial cells (confined by basement membrane, on left side of image) along with numerous interstitial foam cells that are engorged with lipid material (JMS, 400 × original magnification).

Figure 3. Minimal Change Disease. Transmission electron micrograph of peripheral capillary loops demonstrating diffuse effacement of podocyte foot processes (arrowheads). *Glomerular capillary lumen. Left panel 13 300 ×, right panel 17 100 × original magnification. These images were obtained from the same patient as in Figure 1.

Figure 4. Focal Segmental Glomerulosclerosis. Transmission electron micrograph of podocytes exhibiting diffuse effacement of foot processes and prominent microvillous transformation (9420 × original magnification). ‘Microvilli’ (M) are apparent as filamentous projections from the apical portions of podocytes into the urinary space.
Minimal change disease has a favorable prognosis and shows a good response to therapy with corticosteroids, especially in children. In fact, children are given a trial course of corticosteroids when they present with the nephrotic syndrome, without a diagnostic renal biopsy being performed. A proportion of patients experience frequent relapses, especially if the initial relapse occurs within the first 6 months of presentation. A poor response to steroid therapy correlates with an increased risk of progressive kidney disease. Steroid-resistant disease should prompt a kidney biopsy to investigate other etiologies for proteinuria, such as FSGS. Evaluation of genetic causes of minimal change disease/FSGS such as mutations in NPHS1 (nephrin) and NPHS2 (podocin) genes can be performed. The responsiveness of minimal change disease to steroid treatment suggests that it is a disorder of immune regulation. Further evidence to support this hypothesis is the well-known association of Hodgkin lymphoma with minimal change disease. B cell depletion with rituximab can be effective in multirelapsing minimal change disease. However, these effects of rituximab may be independent of its ability to deplete B cells. Rituximab binds to sphingomyelin phosphodiesterase acid-like 3b (SMPDL-3b) protein and regulates acid sphingomyelinase (ASMase) activity in podocytes, which may be cytoprotective in patients at risk for recurrent FSGS after transplant.
Until recently, a spontaneous animal model of minimal change disease was not available. Instead, investigators induced foot process effacement in rats by infusion of puromycin aminonucleoside. An alternative model involved infusion of polycationic protamine sulfate, which neutralizes the surface negative charge of the podocyte’s apical membrane domain and produces rapid foot process effacement. This change is reversible by delivering anionic heparin to neutralize the protamine sulfate. Lastly, lipopolysaccharide (LPS) can be used (albeit in a narrow dose range) to induce foot process effacement in mice. While convenient, these models do not accurately recapitulate the selective proteinuria or glucocorticoid responsiveness often seen in patients with minimal change disease. However, studies of animals injected with LPS identified angiopoeitin-like-4 (Angptl4), a glycoprotein that is secreted in a hyposialylated form by injured podocytes. Clustering of secreted and hyposialylated Angptl4 results in alteration of GBM charge that triggers signaling events in podocytes that lead to foot process effacement. Interestingly, this model recapitulates the glucocorticoid sensitivity and selective proteinuria seen in patients with minimal change disease.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123864567054034
Nephrology
Robert L. Seigle MD, Martin A. Nash MD, in Pediatric Secrets (Fifth Edition), 2011
70 What is the most common form of nephrotic syndrome seen in childhood?
Minimal-change nephrotic syndrome (MCNS). Earlier names for this condition included lipoid nephrosis and nil disease. MCNS is a form of primary nephrotic syndrome and has a more favorable therapeutic response and prognosis. The etiology of MCNS is unknown, but it appears to be a condition of abnormal T-lymphocyte function. Other forms of primary nephrotic syndrome include conditions such as focal segmental glomerulosclerosis, membranous nephropathy, and membranoproliferative glomerulonephritis. Secondary forms of nephrotic syndrome may also occur as a consequence of infection, as a response to some medications, and as an autoimmune phenomenon.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323065610000136
Minimal Change Disease
Howard Trachtman, … Jai Radhakrishnan, in National Kidney Foundation Primer on Kidney Diseases (Sixth Edition), 2014
Terminology and Histopathology
Minimal change disease (MCD) is a common cause of the nephrotic syndrome (NS). Also known as lipoid nephrosis, nil disease, and minimal change nephropathy, the kidney histology on light microscopy in MCD is relatively normal and lacks the significant glomerular cell proliferation, infiltration by circulating immunoeffector cells, immune deposits, tubulointerstitial changes, or alterations in the glomerular basement membrane (GBM) that characterize other glomerular diseases. The defining feature of MCD is diffuse effacement and fusion of podocyte foot processes without electron-dense deposits on electron microscopy. Immunofluorescence is typically negative or may show low-level staining for C3 and IgM.
Although MCD is histologically defined by the previously mentioned criteria, it can also be diagnosed clinically by exhibiting responsiveness to corticosteroid treatment. In children, because MCD is the cause of 90% of cases of idiopathic NS, a kidney biopsy is only warranted if the clinical and laboratory evidence, including disease onset before 6 months of age or following adolescence, unexpected systemic manifestations, or a low serum C3 level, suggests an alternative diagnosis. Children who do not exhibit these characteristics will typically have MCD and will consequently respond to steroids. The nomenclature “steroid-sensitive nephrotic syndrome (SSNS)” is also used to describe such children. Steroid responsiveness is a marker of a favorable long-term prognosis. In contrast, children with steroid-resistant nephrotic syndrome (SRNS) are more likely on subsequent kidney biopsies to show focal segmental glomerulosclerosis (FSGS), a disease that is associated with a worse prognosis. The causes of the NS in adults are more varied and include a higher percentage of cases with membranous nephropathy (MN) and membranoproliferative glomerulonephritis (MPGN). Because MCD only accounts for approximately 10% to 15% of cases, a kidney biopsy is usually warranted to establish the etiology of nephrotic syndrome.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9781455746170000170
The Kidneys and the Urinary System
Ivan Damjanov MD, PhD, in Pathology Secrets (Third Edition), 2009
42 List the typical features of minimal change disease
- ▪
-
It is also known as nil disease (from Latin nihil, “nothing”) and in the clinical setting as lipoid nephrosis.
- ▪
-
It is the most common cause of nephrotic syndrome in children (90% of those younger than 5 years old and 50% of those younger than 10 years old, in comparison with 20% of adults).
- ▪
-
Etiology/pathogenesis is unknown. It is thought to be related to type IV hypersensitivity reaction or the effects of cytokines.
- ▪
-
Pathology: Microscopic and immunofluorescence findings are negative. The glomeruli appear normal.
- ▪
-
Electron microscopy shows reactive fusion of foot processes of epithelial cells.
- ▪
-
Therapy: It responds to corticosteroid treatment but may occasionally recur.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323055949000155
Glomerular Disorders and Nephrotic Syndromes
Gerald B. Appel, Jai Radhakrishnan, in Goldman’s Cecil Medicine (Twenty Fourth Edition), 2012
Diagnosis
In true minimal change disease, histopathology typically reveals no glomerular abnormalities on light microscopy (Fig. 123-1). The tubules may show lipid droplet accumulation from absorbed lipoproteins (hence the older term lipoid nephrosis). Immunofluorescence staining and electron microscopy (Fig. 123-2) show no immune-type deposits. By electron microscopy, the GBM is normal, and effacement or “fusion” of the visceral epithelial foot processes is noted along virtually the entire distribution of every capillary loop.
Treatment
The course of minimal change nephrotic syndrome is often one of remissions, relapses, and responses to additional treatment. When treated with corticosteroids for 8 weeks, 85 to 95% of children experience a remission of proteinuria. In adults, the response rate is somewhat lower, with 75 to 85% of patients responding to regimens of daily (1 mg/kg, maximum 80 mg) or alternate-day (2 mg/kg, maximum 120 mg) prednisone therapy, tapered after 2 months of treatment for a total of 5 to 6 months of therapy. The time to clinical response is slower in adults, and they are not considered steroid resistant until they have failed to respond to 16 weeks of treatment. Tapering of the steroid dose after remission should be gradual over 1 to 2 months. Approximately 40% of adults relapse by 1 year and 50% by 5 years. Most clinicians treat the first relapse similarly to the initial episode. Patients who relapse a third time or who become corticosteroid dependent (unable to decrease the prednisone dose without proteinuria recurring) may be treated with a 2- to 3-month course of the alkylating agent cyclophosphamide at a dose of up to 2 mg/kg/day. Up to 50% of patients will have a remission of at least 5 years, but the response rate is lower in corticosteroid-dependent patients. Other alternative treatments for patients who frequently relapse or are steroid resistant include low-dose cyclosporine (3 to 5 mg/kg/day in divided doses for 4 months), tacrolimus (0.05 to 0.1 mg/kg/day in divided doses), and mycophenolate mofetil (750 to 1000 mg twice daily). All provide approximately equivalent remission and relapse rates, but cyclosporine and tacrolimus increase the risk of nephrotoxicity, so their blood levels must be carefully monitored.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9781437716047001238
Role of Electron Microscopy in Modern Diagnostic Surgical Pathology
ROBERT A. ERLANDSON, in Modern Surgical Pathology (Second Edition), 2009
Glomerulopathies
The most common use of electron microscopy is for the evaluation of glomerulopathies. It is customary to divide percutaneous renal biopsies obtained from patients with nephrotic syndrome for light microscopic (H&E, periodic acid-Schiff [PAS], silver methenamine, and trichrome stains), immunofluorescent, and ultrastructural studies, because establishing a correct diagnosis has therapeutic and prognostic implications.20,21 The electron microscope is invaluable for resolving the constituents of the glomerulus, including the glomerular basement membrane (GBM), which ranges in thickness from 150 nm at birth to approximately 300 nm to 400 nm in adulthood; the blood capillary endothelial cell; the foot processes of the visceral epithelial cell (podocyte), with their “filtration slit pores” that line the urinary space; the mesangium (axial region of the glomerulus); and Bowman’s capsule, which is lined on its inner surface by flattened parietal epithelial cells.
Primary and secondary (systemic) glomerular diseases in which ultrastructural studies are crucial to an accurate diagnosis include (1) minimal change nephritic syndrome (also called nil disease or lipoid nephrosis), which is characterized by extensive fusion of podocyte foot processes; (2) benign familial recurrent hematuria with marked thinning of the GBM (<180 nm in children; <250 nm in adults), also called thin basement membrane disease; (3) Alport’s syndrome (a hereditary glomerulopathy), which results in marked thickening, attenuation, and lamellarization of the GBM (a result of injury and repair); and (4) Berger’s disease (immunoglobulin A [IgA] nephropathy), exhibiting focal splitting and disruption of the GBM as well as finely granular mesangial deposits. Electron microscopy is also helpful for visualizing small peri-GBM, GBM, and mesangial deposits in, for example, early (stage 1) membranous glomerulonephritis, in which only scattered, small, subepithelial, dense immunoglobulin or light-chain deposits are evident (Fig. 6-1), and thrombotic microangiopathy (hemolytic uremic syndrome), with subendothelial deposits of an electron-lucent substance and microfibrils, formation of a thin endothelial basement membrane, mesangial interposition, and intraluminal (capillary) thrombi.
More recently, a number of glomerulopathies with organized deposits have been recognized.22 These include the rare fibrillar, immunotactoid, and cryoglobulinemic glomerulopathies and the more common amyloidosis. The differential diagnosis of the three rare types depends on the ultrastructural identification of disease-specific glomerular deposits. The pertinent diagnostic features (primarily ultrastructural) of these diseases can be summarized as follows. Amyloidosis (proteinuria-nephrotic syndrome) often occurs in myeloma (plasma cell dyscrasia) patients; the usual type found is AL amyloidosis (abnormal light chains are present). Ultrastructurally, numerous 8- to 12-nm nonbranching fibrils are randomly distributed in the glomerulus. Fibrillary glomerulopathy is characterized by a ribbon-like pattern of IgA and C3 and a primarily subepithelial deposition of 20- to 30-nm fibrils with an amyloid P component. In immunotactoid glomerulopathy, IgG, C3, and characteristic clusters of spherical microtubular structures with a 30- to 40-nm diameter are also found in the mesangium. The cryoglobulinemic glomerulopathies are divided into types I, II, and III and are associated with B-cell lymphoplasmacytoid malignancies. Type I is identified by IgG and 80-nm-wide bundles of rigid fibrils or “fingerprint” arrays of tubular structures, all of which are distributed throughout the glomerulus. In types II and III, mixed cryoglobulins and 25-nm microtubules are found in thrombi and in the subepithelial and mesangial regions of the glomerulus. For more detailed information on all the glomerulopathies, see Chapter 29 and the cited references.20-23
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9781416039662000059
Nephrotic Syndrome
Ayesa N. Mian MD, in Pediatric Clinical Advisor (Second Edition), 2007
Basic Information
Definition
Nephrotic syndrome (NS) is characterized by proteinuria (>40 mg/m2/hr), hypoalbuminemia (<2.5 g/dL), edema, and hypercholesterolemia. Primary NS is a disease involving only the kidney, and it is not associated with extrarenal manifestations. Secondary NS occurs as a manifestation of systemic disease that involves the kidney, such as systemic lupus erythematosus (SLE), Henoch‐Schönlein purpura (HSP), sickle cell anemia, or uncommonly acute poststreptococcal glomerulonephritis. Primary NS is addressed here.
Synonyms
-
Steroid‐resistant nephrotic syndrome
-
Diffuse mesangial proliferation
-
Focal‐segmental glomerulosclerosis (FSGS)
-
Steroid‐sensitive nephrotic syndrome
-
Idiopathic nephrotic syndrome
-
Lipoid nephrosis
-
Minimal change disease
-
Nil disease
ICD‐9‐CM Codes
-
581.1 Focal segmental glomerulosclerosis (FSGS)
-
581.1 Membranous glomerulonephritis
-
581.2 Membranoproliferative glomerulonephritis (MPGN)
-
581.3 Minimal change nephrotic syndrome (MCNS)
-
581.9 Nephrotic syndrome, not otherwise specified
Epidemiology & Demographics
- •
-
Age
- ○
-
NS affects children of all ages.
- ○
-
NS is rare in the first year of life.
- ○
-
Congenital NS typically manifests in the first 3 months of life.
- •
-
Steroid‐sensitive nephrotic syndrome
- ○
-
Incidence varies with race, ethnicity, and age.
- ○
-
Incidence is estimated at approximately 2 to 2.7 cases per 100,000 children in the United States.
- ○
-
Cumulative U.S. prevalence is estimated to be 16 cases per 100,000 children.
- •
-
Gender
- ○
-
In young children, the male‐to‐female ratio is 2:1.
- ○
-
In adolescents, the male‐to‐female ratio is 1:1.
- •
-
MCNS accounts for 60% to 90% of NS cases among children and most cases among children younger than 7 years.
- •
-
In older children, other diagnoses become more prevalent.
- •
-
Genetics: increased incidence among family members
- •
-
FSGS
- ○
-
FSGS accounts for approximately 10% of children with NS.
- ○
-
The prevalence of FSGS may be increasing.
- ○
-
Approximately 20% of patients are steroid sensitive.
Clinical Presentation
History
- •
-
Recent history of upper respiratory infection or other viral illness
- •
-
History of allergies
- •
-
Edema that is dependent
- ○
-
Insidious onset; often first noticed in the periorbital region in the morning
- ○
-
May be present for weeks
- ○
-
Often attributed to allergies
- ○
-
Progresses to lower extremities, abdomen, and can become generalized (i.e., anasarca)
- •
-
Weight gain
- •
-
Changes in urination
- ○
-
Decreased urine output
- ○
-
Gross hematuria unusual with MCNS
- •
-
Respiratory difficulty
- ○
-
Suggests pulmonary edema, pleural effusions, or significant abdominal distention
- •
-
Anorexia
- •
-
Diarrhea resulting from edema of the intestinal wall
- •
-
Vomiting
- •
-
Abdominal pain
- ○
-
May occur with peritonitis and be associated with fever
- ○
-
May occur with hypovolemia
- ○
-
May occur with rapid development of ascites
- ○
-
May occur with thrombosis
- •
-
Renal vein thrombosis is associated with gross hematuria and flank pain.
- •
-
Headaches
- ○
-
May suggest associated hypertension
Physical Examination
- •
-
Signs of volume overload
- ○
-
Periorbital edema
- ○
-
Decreased breath sounds, which suggest pleural effusions
- ○
-
Rales
- ○
-
Ascites
- ○
-
Presacral edema, scrotal or labial edema
- ○
-
Peripheral edema
- ○
-
Facial edema (difficult to assess when first seeing the child; often helpful to compare the current appearance with a photograph taken before the illness)
- •
-
Signs of intravascular volume depletion
- ○
-
Tachycardia
- ○
-
Hypotension
- ○
-
Poor perfusion
- ○
-
Dry mucous membranes
- ○
-
Orthostatic changes
- •
-
Signs of peritonitis
- ○
-
Fever
- ○
-
Abdominal pain
- ○
-
Distention
- ○
-
Rebound
- ○
-
Guarding
- ○
-
Tenderness
- ○
-
Absence of bowel sounds
- •
-
Signs of skin breakdown
- ○
-
Infection can spread quickly in nephrotic children.
- •
-
Rash or joint swelling
- ○
-
May suggest that the disease is a secondary form of NS
Etiology
- •
-
The cause of primary NS is unknown, but evidence suggests abnormal T‐cell function in MCNS.
- ○
-
Response to immunomodulatory medications, such as prednisone
- ○
-
Relapses often associated with upper respiratory and other minor illness
- ○
-
Improves after measles
- ○
-
Increased frequency of atopy and allergies in children with MCNS
- ○
-
Associated with Hodgkin’s lymphoma and non‐Hodgkin’s lymphoma
- •
-
Secondary NS occurrs as a complication of other glomerular insults
- ○
-
SLE
- ○
-
HSP
- ○
-
Sickle cell anemia
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323035064102251
Nephrology
Sharma S. Prabhakar M.D., M.B.A., F.A.C.P., F.A.S.N., in Medical Secrets (Fifth Edition), 2012
Proteinuria, nephrotic syndrome, and nephritic syndrome
16 List the four general mechanisms by which abnormally increased urinary protein excretion (>150 mg/day) occurs.
- ▪
-
Glomerular
- ▪
-
Tubular
- ▪
-
Overflow
- ▪
-
Secretory
17 What causes glomerular proteinuria?
Damage to the glomerular filtration barrier (in glomerulonephritis), leading to leakage of plasma proteins into the glomerular ultrafiltrate.
18 Describe the mechanism behind tubular proteinuria.
Suboptimal reabsorption of the normally filtered protein as a result of tubular disease. This recovery of the small amount of normally filtered protein (usually ~2 g/day) allows for the normal excretion of < 150 mg/day of protein.
19 Explain overflow proteinuria.
Proteinuria resulting from disease states that lead to excessive levels of plasma proteins (e.g., in multiple myeloma). The proteins are filtered and overload the reabsorptive capacity of the renal tubules.
20 What is secretory proteinuria?
Proteinuria that occurs because of the addition of protein to the urine after glomerular filtration. The protein may come from the renal tubules (e.g., Tamm-Horsfall protein from the ascending limb of the loop of Henle) or from the lower genitourinary (GU) tract.
21 What conditions are associated with heavy proteinuria despite severely reduced GFR?
Usually glomerular disease. In most glomerular diseases, proteinuria tends to decrease with diminishing GFR as the filtration of proteins also tends to decrease. However, in certain conditions, such as diabetic nephropathy, amyloidosis, focal glomerulosclerosis, and probably reflex nephropathy, proteinuria (often in the nephrotic range) persists despite severely diminished GFR.
22 Define “nephrotic syndrome.”
A symptom complex resulting from various etiologies and characterized by heavy proteinuria (usually >3.5 g/day), generalized edema, and lipiduria with hyperlipidemia. Because all the other features are a consequence of marked proteinuria, some authorities restrict the definition of “nephrosis” to heavy proteinuria alone.
23 What are the common causes of nephrotic syndrome in adults and children?
In adults, the most common cause is diabetes nephropathy, which is a secondary cause of nephritic syndrome. Membranous nephropathy is the most common primary glomerulopathy in adults. In children, the most common cause of nephrotic syndrome is minimal change disease, also called “lipoid nephrosis” or “nil disease.” Other causes of nephrotic syndrome include focal and segmental glomerulosclerosis and amyloidosis.
24 When evaluating patients with nephrotic syndrome, which diseases must you rule out before considering the syndrome to be due to a primary renal disease?
- ▪
-
Drugs that may result in excessive urinary protein excretion (gold and penicillamine)
- ▪
-
Systemic infections (hepatitis B and C, human immunodeficiency virus [HIV], malaria)
- ▪
-
Neoplasia (lymphomas)
- ▪
-
Multisystem collagen vascular diseases (systemic lupus erythematosus [SLE])
- ▪
-
Diabetes mellitus (DM)
- ▪
-
Heredofamilial diseases (Alport’s syndrome)
25 Why is it important to distinguish primary renal disease from these conditions?
The distinction between these causes and primary renal disease is important for a number of reasons. Diagnostically, identification of some of these processes may help to identify the renal lesion without the need for a renal biopsy (as in DM). Treatment of such disorders may involve simple discontinuation of the offending agent (e.g., a drug). Management may need to be directed at a systemic disease (infection) rather than at the renal lesion itself.
26 Name the common complications of the nephrotic syndrome.
- ▪
-
Edema and anasarca.
- ▪
-
Hypovolemia with acute prerenal or parenchymal renal disease or both. In the nephrotic syndrome, decreased effective arterial blood volume can lead to various degrees of renal underperfusion, resulting in renal failure in severe cases.
- ▪
-
Protein malnutrition due to massive protein losses in excess of dietary replacement.
- ▪
-
Hyperlipidemia, which raises the risk of atherosclerotic cardiovascular disease.
- ▪
-
Increased susceptibility to bacterial infection often involving the lungs, meninges (meningitis), and peritoneum. Common organisms include Streptococcus (including Streptococcus pneumoniae), Haemophilus influenzae, and Klebsiella spp.
- ▪
-
Proximal tubular dysfunction leading to Fanconi’s syndrome with urinary wasting of glucose, phosphate, amino acids, uric acid, potassium, and bicarbonate.
- ▪
-
Hypercoagulable state manifested by an increased incidence of venous thrombosis, particularly in the renal vein, which may be due to urinary loss of antithrombotic factors.
27 Define “nephritic syndrome.”
A renal disorder resulting from diffuse glomerular inflammation characterized by the sudden onset of gross or microscopic hematuria, decreased GFR, oliguria, hypertension (HTN), and edema. Nephritic syndrome results from many different etiologies but is traditionally represented by postinfectious glomerulonephritis following infections with certain strains of group A beta-hemolytic streptococci.
28 What are the various causes of an acute nephritic syndrome?
- ▪
-
Postinfectious glomerulonephritis: bacterial (pneumococci, Klebsiella spp., staphylococci, gram-negative rods, and meningococci), viral (varicella, infectious mononucleosis, mumps, measles, hepatitis B, and coxsackievirus), rickettsial (Rocky Mountain spotted fever, and typhus), and parasitic (e.g., Falciparum malaria, toxoplasmosis, and trichinosis)
- ▪
-
Idiopathic glomerular diseases: membranoproliferative glomerulonephritis, mesangial proliferative glomerulonephritis, and immunoglobulin A (IgA) nephropathy
- ▪
-
Multisystem diseases: SLE, Henoch-Schönlein purpura, essential mixed cryoglobulinemia, and infective endocarditis
- ▪
-
Miscellaneous: Guillain-Barré syndrome and post-irradiation of renal tumors
29 Are the syndromes of nephritis and nephrosis mutually exclusive?
No. Some forms of glomerular diseases are characteristically nephrotic in their presentation whereas some aggressive forms of proliferative glomerulopathies present as nephritic syndrome. Some others manifest mixed features (Table 8-1).
30 A 62-year-old man with nephrotic syndrome is found to have no systemic etiology. What is the differential diagnosis?
As opposed to minimal lesion in children, minimal lesion on renal biopsy in an elderly patient warrants an extensive search to rule out underlying malignancy, especially lymphomas (both Hodgkin’s and non-Hodgkin’s) and other solid tumors (e.g., renal cell carcinoma). One third of elderly patients with membranous nephropathy have underlying malignancy (colon, stomach, or breast).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323063982000096
ГЛОМЕРУЛОНЕФРИТ
ХАРАКТЕРИЗУЕТСЯ ВОСПАЛЕНИЕМ канальцев
интерстиция почечных лоханок почечных
клубочков
КОМПОНЕНТЫ
ЮКСТАГЛОМЕРУЛЯРНОГО КОМПЛЕКСА плотное
пятно клетки Гурмагтига мезангиальные
клетки васкулярные клетки
юкстагломерулярные
клетки
|
Установить |
||
|
3. |
ХАРАКТЕРИСТИКА |
|
|
1) |
подоциты |
|
|
2) |
мезангиальные |
А) |
|
3) |
эндотелиальные |
Б) |
|
4) |
интерстициальные |
В) |
|
диафрагмы |
||
|
5) |
юкстагломерулярные |
Г) |
|
ренин |
||
|
Д) |
выполняют
сократительную функцию
Ответ:
1-в
, 2-д
, 3-б
, 4-а
, 5-г
.
|
4. |
||
|
ТЕРМИН |
СОДЕРЖАНИЕ |
|
|
1) |
анурия |
А) |
|
2) |
олигурия |
Б) |
|
3) |
полиурия |
В) |
|
4) |
уролитиаз |
Г) |
|
5) |
протеинурия |
Д) |
|
6) |
Е) |
|
|
Ж) |
кислоты
в крови Ответ: 1-е
, 2-д
, 3-в
, 4-а
, 5-ж(по
идее это ответ 6,но в ответах так) , 6- .
5.
ОСНОВНОЕ ПРОЯВЛЕНИЕ БОЛЕЗНИ МИНИМАЛЬНЫХ
ИЗМЕНЕНИЙ отек стромы пролиферация
мезангиоцитов
склероз
и гиалиноз базальной мембраны исчезновение
ножек отростков подоцитов инфильтрация
нейтрофилами
|
6. |
|||
|
ПОРАЖЕНИЕ |
ЛОКАЛИЗАЦИЯ |
||
|
1) |
фокальное |
А) |
|
|
2) |
диффузное |
Б) |
|
|
3) |
глобальное |
В) |
|
|
4) |
сегментарное |
Г) |
|
|
Ответ: |
|||
|
7.Установить |
|||
|
ТИП |
КЛИНИЧЕСКИЙ |
||
|
ИЗМЕНЕНИЙ |
|||
|
1) |
утолщение |
А) |
|
|
2) |
некроз |
Б) |
|
|
канальцев |
|||
|
3) |
пролиферация |
В) |
|
|
или |
|||
|
4) |
клеточная |
Г) |
|
|
повреждение |
недостаточность |
||
|
Д) |
|||
|
нефритический |
|||
|
Ответ: |
ОСНОВНОЙ
МЕХАНИЗМ ПАТОГЕНЕЗА БОЛЬШИНСТВА ФОРМ
ГЛОМЕРУЛОНЕФРИТА иммунные комплексы
in situ
циркулирующие
иммунные комплексы
КОМПОНЕНТЫ
КОМПЛЕМЕНТА ВЫЗЫВАЮТ
адгезию
лейкоцитов гемодинамические
нарушения пролиферацию
мезангиальных клеток
накопление внеклеточного матрикса
НЕФРИТОГЕННЫМИ
СВОЙСТВАМИ ОБЛАДАЕТ золотистый
стафиликокк бета-гемолитический
стрептококк группы А
МОРФОЛОГИЧЕСКАЯ
ХАРАКТЕРИСТИКА
ОСТРОГО
ГЛОМЕРУЛОНЕФРИТА субэпителиальные
депозиты потеря
отростков ножек подоцитов
пролиферация
мезангиальных клеток нейтрофилы в
просветах капиллярных петель пролиферация
и набухание эндотелиальных клеток
выраженная
лимфогистиоцитарная инфильтрация
капиллярных
петель
АНТИГЕНАМИ
ПРИ ОСТРОМ ПОСТСТРЕПТОКОККОВОМ
ГЛОМЕРУЛОНЕФРИТЕ
ЯВЛЯЮТСЯ
базальные мембраны канальцев
гломерулярные
базальные мембраны поврежденные
эпителиальные клетки
поврежденные
стрептококковым ферментом иммуноглобулины
ПРОЯВЛЕНИЕМ
КОАГУЛОПАТИИ ПРИ ОСТРОМ ПОСТСТРЕПТОКОККОВОМ
ГЛОМЕРУЛОНЕФРИТЕ
ЯВЛЯЮТСЯ депозиты
фибрина гиалиновые тромбы гиалиновые
цилиндры лейкоцитарная инфильтрация
пролиферация
и набухание интерстициальных клеток
ГЛОМЕРУЛОНЕФРИТЫ
ПРОТЕКАЮЩИЕ В КЛИНИКЕ ОСТРО
быстро
прогрессирующий подострый
мембранопролиферативный
острый постстрептококковый
фокальный
пролиферативный и некротизирующий
ВАРИАНТЫ
БЫСТРО ПРОГРЕССИРУЮЩЕГО ГЛОМЕРУЛОНЕФРИТА
идиопатический
постинфекционый при
паразитарных инфекциях
при
системных заболеваниях
БЫСТРО
ПРОГРЕССИРУЮЩИЙ ГЛОМЕРУЛОНЕФРИТ ПРИ
СИНДРОМЕ ГУДПАСЧЕРА
ПО
МЕХАНИЗМУ РАЗВИТИЯ антительны
иммунокомплексный
ОСТРЫЙ
ПОСТСТРЕПТОКОККОВЫЙ ГЛОМЕРУЛОНЕФРИТ
СОПРОВОЖДАЕТСЯ удвоением ГБМ формированием
полулуний
развитием
гиперклеточности клубочков
ПРИ
ОСТРОМ ГЛОМЕРУЛОНЕФРИТЕ ПОЧКИ
плотные
с сальным блеском
увеличены
в размерах 34
с петехиальными кровоизлияниями на
поверхности
ПОЛУЛУНИЯ
ПРИ БЫСТРО ПРОГРЕССИРУЮЩЕМ ГЛОМЕРУЛОНЕФРИТЕ
ОБРАЗУЮТСЯ
В РЕЗУЛЬТАТЕ набухания эндотелиальных
клеток миграции плазмоцитов и нейтрофилов
пролиферации
париетальных клеток пролиферации
мезангиальных клеток
ПОЛУЛУНИЯ
ПРИ БЫСТРО ПРОГРЕССИРУЮЩЕМ ГЛОМЕРУЛОНЕФРИТЕ
СО
ВРЕМЕНЕМ
ПОДВЕРГАЮТСЯ атрофии склерозу
рассасыванию
петрификации
К
ПОСТСТРЕПТОКОККОВОМУ ГЛОМЕРУЛОНЕФРИТУ
ОТНОСЯТ лобулярный
быстро
прогрессирующий острый
пролиферативный мембранопролиферативный
22.
СИМПТОМЫ НЕФРОТИЧЕСКОГО СИНДРОМА
-
1)
гиперурикемия
4)
массивная
протеинурия2)
гиперлипидемия
5)
генерализованные
отеки3)
гипоальбуминемия
6)
массивная
макрогематурия
Установить
правильную последовательность.
23.
ЗВЕНЬЯ НЕФРОТИЧЕСКОГО СИНДРОМА
3-отеки
2-альбуминурия
гиперальбуминурия
1-усиление
гломерулярной проницаемости усиление
канальцевого катаболизма белков
Установить
правильную последовательность.
24.
ЭТАПЫ РАЗВИТИЯ ТРОМБОТИЧЕСКИХ ОСЛОЖНЕНИЙ
ПРИ НЕФРОТИЧЕСКОМ
СИНДРОМЕ
5-тромбоз
3-гиперлипидемия 2-гиперальбуминурия
4-усиление
агрегации тромбоцитов усиление
синтеза нуклеопопротеинов 1-усиление
гломерулярной проницаемости
Установить
правильную последовательность.
ЭТАПЫ
РАЗВИТИЯ ВТОРИЧНЫХ ИНФЕКЦИЙ
ПРИ
НЕФРОТИЧЕСКОМ СИНДРОМЕ 4-развитие
инфекций 3-ослабление иммунитета 2-потеря
иммуноглобулинов
1-усиление
гломерулярной проницаемости
НЕФРОТИЧЕСКИЙ
СИНДРОМ У ДЕТЕЙ ОБЫЧНО РАЗВИВАЕТСЯ ПРИ
системных болезнях накопления первичном
поражении почек
НЕФРОТИЧЕСКИЙ
СИНДРОМ ЧАСТО ОСЛОЖНЯЕТ ТЕЧЕНИЕ
СКВ
Амилоидоза
склеромы
болезни Рейно
несахарного
диабета
ПЕРВИЧНЫЕ
ЗАБОЛЕВАНИЯ ПОЧЕК, СОПРОВОЖДАЮЩИЕСЯ
ЧАСТО
НЕФРОТИЧЕСКИМ
СИНДРОМОМ Ig-A нефропатия
липоидный
нефроз мембранозная нефропатия
мембранопролиферативный
гломерулонефрит
«ШИПИКИ»
ПРИ МЕМБРАНОЗНОЙ НЕФРОПАТИИ ВЫЯВЛЯЮТСЯ
ПРИ ОКРАСКЕ
|
1) |
суданом |
4) |
пикрофуксином |
|
2) |
серебрением |
5) |
толуидиновым |
|
3) |
конго-красным |
6) |
гематоксилином |
Установить
правильную последовательность.
МОРФОЛОГИЧЕСКИЕ
ПРОЯВЛЕНИЯ МЕМБРАНОЗНОЙ НЕФРОПАТИИ
1-появление
«шипиков» 2-диффузное утолщение ГБМ
3-субэпителиальное
отложение иммунных депозитов
4-погружение
иммунных депозитов в базальную мембрану
При
болезни минимальных изменений
гистологически клубочки
изменены
не
изменены
СИНОНИМ
БОЛЕЗНИ МИНИМАЛЬНЫХ ИЗМЕНЕНИЙ – НЕФРОЗ
некротический желтушный липоидный
болезнь
малых отростков подоцитов
КЛИНИЧЕСКАЯ
ХАРАКТЕРИСТИКА ИДИОПАТИЧЕСКОГО
ФОКАЛЬНОГО
СЕГМЕНТАРНОГО
ГЛОМЕРУЛОСКЛЕРОЗА неблагоприятный
прогноз неселективная
гематурия и гипертензия
плохой
ответ на кортикостроидную терапию
развитие в финале хронической почечной
недостаточности
ГИСТОЛОГИЧЕСКИЕ
ВАРИАНТЫ ФОКАЛЬНОГО СЕГМЕНТАРНОГО
ГЛОМЕРУЛОСКЛЕРОЗА
концевой
клеточный
мембранозная
нефропатия
коллабирующая
гломерулопатия
ГИСТОЛОГИЧЕСКАЯ
ХАРАКТЕРИСТИКА ФОКАЛЬНОГО СЕГМЕНТАРНОГО
ГЛОМЕРУЛОСКЛЕРОЗА
гиперклеточность клубочков склероз
сегментов клубочков накопление гиалиновых
масс увеличение
мезангиального матрикса
коллапс
капиллярных петель клубочков диффузное
утолщение базальных мембран капилляров
клубочков
ДИФФУЗНОЕ
УТОЛЩЕНИЕ ГЛОМЕРУЛЯРНЫХ БАЗАЛЬНЫХ
МЕМБРАН ПРИ
МЕМБРАНОЗНОЙ
НЕФРОПРАТИИ ВЫЯВЛЯЮТ ПРИ ОКРАСКЕ
-
1)
суданом
III5)
пикрофуксином
2)
фукселином
6)
толуидиновым
синим3)
серебрением
7)
шифф-иодной
кислотой4)
конго-красным

гематоксилином
и эозином

37.
ВАРИАНТЫ МЕМБРАНОПРОЛИФЕРАТИВНОГО
ГЛОМЕРУЛОНЕФРИТА
плотными
депозитами фокальный
гломерулонефрит
субэпителиальными
депозитами
субэндотелиальными
депозитами мезангиопролиферативный
гломерулонефрит экстракапиллярный
пролиферативный гломерулонефрит
ИНТЕРПОЗИЦИЯ
МЕЗАНГИЯ ПРИ МЕМБРАНОПРОЛИФЕРАТИВНОМ
ГЛОМЕРУЛОНЕФРИТЕ ПРИВОДИТ К (ОТВЕТ
1,2,3,5?!)
удвоению
гломерулярных базальных мембран склерозу
и гиалинозу мезангиального матрикса
диффузному
утолщению гломерулярных базальных
мембран
ГИСТОЛОГИЧЕСКИЕ
ИЗМЕНЕНИЯ КЛУБОЧКОВ ПРИ
МЕМБРАНОПРОЛИФЕРАТИВНОМ
ГЛОМЕРУЛОНЕФРИТЕ дольчатый
вид склероз сосудистых петель
многоклеточность
наличие экставазатов
расширение
мезангиального матрикса капли гликогена
в мезангиальном матриксе
40.
ВТОРИЧНЫЙ МЕМБРАНОПРОЛИФЕРАТИВНЫЙ
ГЛОМЕРУЛОНЕФРИТ РАЗВИВАЕТСЯ
ПРИ
СКВ
гепатите В гепатите
С
синдроме
Шегрена сыпном тифе
41.
СИНОНИМ БОЛЕЗНИ БЕРЖЕ – НЕФРОПАТИЯ
|
IgA |
|
|
IgM |
1 |
|
IgG |
ГЛАВНЫЙ
КЛИНИЧЕСКИЙ СИМПТОМ IGA-НЕФРОПАТИИ отеки
тяжелая
протеинурия
рецидивирующая
макро- и микрогематурия
IGA-НЕФРОПАТИЯ
ВКЛЮЧЕНА В ГРУППУ мезангиопролиферативного
гломерулонефрита мембранопролиферативного
гломерулонефрита фокального сегментарного
гломерулярного склероза
ХРОНИЧЕСКИЙ
ГЛОМЕРУЛОНЕФРИТ В ИСХОДЕ ПОСТСТРЕПТОКОККОВОГО
ГЛОМЕРУЛОНЕФРИТА
РАЗВИВАЕТСЯ редко часто
МАКРОСКОПИЧЕСКАЯ
ХАРАКТЕРИСТИКА ПОЧЕК
ПРИ
ХРОНИЧЕСКОМ ГЛОМЕРУЛОНЕФРИТЕ увеличены
симметрично
сморщены
мелкозернистая
поверхность истончение коркового
вещества
отложение
жировой ткани вокруг лоханок
46.
К ВНЕПОЧЕЧНЫМ ИЗМЕНЕНИЯМ ПРИ ХРОНИЧЕСКОМ
ГЛОМЕРУЛОНЕФРИТЕ
ОТНОСЯТ
УРЕМИЧЕСКИЙ гепатит
тиреоидит простатит
перикардит
гастроэнтерит
47.
ДИАЛИЗНЫЕ ИЗМЕНЕНИЯ В ПОЧКАХ ПРИ
ТЕРМИНАЛЬНОЙ СТАДИИ
ХРОНИЧЕСКОГО
ГЛОМЕРУЛОНЕФРИТА ВКЛЮЧАЮТ болезнь
Берже кистозная болезнь почек
кальцификация
в почечном клубочке фибриноидный
некроз стенок артерий утолщение
внутренней оболочки артерий
|
Установить |
||
|
48. |
МОРФОЛОГИЧЕСКИЙ |
|
|
ПРОЛИФЕРАТИВНОГО |
ПРИЗНАК |
|
|
ГЛОМЕРУЛОНЕФРИТА |
||
|
1) |
I |
А) |
|
2) |
II |
Б) |
|
3) |
III |
В) |
|
Г) |
||
|
Ответ: |
49.
МЕТОД МОРФОЛОГИЧЕСКОЙ ДИАГНОСТИКИ
БОЛЕЗНИ
БЕРЖЕ иммунофлуоресцентный
электронная
микроскопия селективная гистологическая
окраска
Установить
правильную последовательность.
50.
ПАТОГЕНЕЗ IGA-НЕФРОПАТИИ
1-отложение
IgA-депозитов в мезангии гломерул
2-увеличение
синтеза IgA слизистыми оболочками
врожденная
или приобретенная аномалия
ретикулоэндотелиальной системы
3-увеличение
содержания полимерного IgA и циркулирующих
иммунных комплексов IgA в сыворотке крови
4-экспозиция в дыхательной системе или
желудочно-кишечном тракте провоцирующих
факторов окружающей среды
ГИСТОЛОГИЧЕСКИЕ
ВАРИАНТЫ ГЛОМЕРУЛЯРНЫХ ИЗМЕНЕНИЙ ПРИ
СКВ IgA-нефропатия
фокальный
гломерулонефрит диффузный мембранозный
гломерулонефрит
мезангиопролиферативный
гломерулонефрит диффузный пролиферативный
гломерулонефрит экстракапиллярный
пролиферативный гломерулонефрит
МЕХАНИЗМ
РАЗВИТИЯ ГЛОМЕРУЛОНЕФРИТА
ПРИ
БАКТЕРИАЛЬНОМ ЭНДОКАРДИТЕ антительный
иммунокомплексный
ПРИ
НЕКРОЗЕ ЭПИТЕЛИЯ ИЗВИТЫХ КАНАЛЬЦЕВ
СМЕРТЬ БОЛЬНОГО МОЖЕТ
НАСТУПИТЬ
ОТ ПОЧЕЧНОЙ НЕДОСТАТОЧНОСТИ острой
подострой
хронической
ЗАБОЛЕВАНИЯ,
СОПРОВОЖДАЮЩИЕСЯ ПОВРЕЖДЕНИЕМ КЛУБОЧКОВ
ПОЧЕК
СКВ
синдром
Шегрена болезнь
Шенлейна-Геноха бактериальный эндокардит
тромбоцитопеничееская
пурпура
55.
ПАТОГЕНЕЗ ГЛОМЕРУЛОСКЛЕРОЗА ПРИ САХАРНОМ
ДИАБЕТЕ СВЯЗАН С
ДИАБЕТИЧЕСКОЙ
макроангиопатией
микроангиопатией
Установить
правильную последовательность.
ПАТОГЕНЕЗ
ДИАБЕТИЧЕСКОЙ НЕФРОПАТИИ 1-гломерулосклероз
кистозная
трансформация клубочков
2-усиление
процесса гликозилирования конечных
продуктов
3-утолщение
ГБМ, увеличение синтеза мезангиального
матрикса
4-метаболические
нарушения, связанные с недостаточностью
инсулина
МОРФОЛОГИЧЕСКИЕ
ИЗМЕНЕНИЯ В КЛУБОЧКАХ
ПРИ
ДИАБЕТИЧЕСКОЙ НЕФРОПАТИИ удвоение ГБМ
утолщение
ГБМ эпителиальные
полулуния
узелковый
гломерулосклероз диффузный гломерулосклероз
58.
ДИФФУЗНЫЙ ГЛОМЕРУЛОСКЛЕРОЗ
ПРИ
ДИАБЕТИЧЕСКОЙ НЕФРОПАТИИ ПРИВОДИТ К
облитерирующему
гломерулосклерозу фокальному
сегментарному гломерулярному гиалинозу
СРЕДИ
ПЕРЕЧИСЛЕННЫХ ТЕРМИНОВ ОТМЕТИТЬ СИНОНИМЫ
синдром Гудпасчера узелковый
гломерулосклероз
синдром
Киммельстиля-Уилсона болезнь
минимальных изменений интеркапиллярный
гломерулосклероз
МАКРОСКОПИЧЕСКАЯ
ХАРАКТЕРИСТИКА ПОЧЕК
|
ПРИ |
|||
|
1) |
белый |
4) |
гладкая |
|
2) |
уменьшены |
5) |
ломкая |
|
3) |
увеличены |
6) |
резко |
Установить
правильную последовательность.
СТАДИИ
АМИЛОИДОЗА ПОЧЕК 1-латентная
4-уремическая 3-нефротическая
2-протеинурическая
гипертоническая
НАИБОЛЕЕ
ЧАСТЫЙ ВАРИАНТ АМИЛОИДОЗА ПОЧЕК первичный
старческий вторичный
локальный
ЭЛЕКТИВНАЯ
ОКРАСКА ДЛЯ ВЫЯВЛЕНИЯ
МАСС
АМИЛОИДА судан III
фукселин
пикрофуксин
конго
красный
ДЛЯ
ДИАГНОСТИКИ АМИЛОИДОЗА В СЕКЦИОННОМ
ЗАЛЕ ИСПОЛЬЗУЮТСЯ теллурит калия соли
тетразолия раствор
Люголя 10% серная кислота
|
Установить |
||
|
65. |
МЕХАНИЗМ |
|
|
1) |
синдром |
А) |
|
2) |
синдром |
Б) |
|
3) |
гранулематоз |
В) |
|
4) |
болезнь |
Г) |
|
антител |
||
|
5) |
болезнь |
Д) |
|
цепь |
||
|
Отвеат: |
ПРИ
СИНДРОМЕ ГУДПАСЧЕРА ПОРАЖАЮТСЯ почки
и печень почки
и легкие почки
и сердце
ПЛАЗМОКЛЕТОЧНЫЕ
ДИСКРАЗИИ, ВЫЗЫВАЮЩИЕ ИЗМЕНЕНИЯ В
ПОЧЕЧНЫХ
КЛУБОЧКАХ
болезнь Вальденстрема
множественная
миелома нефропатия легких цепей болезнь
тяжелых цепей Франклина
НЕФРИТ
ПРИ СИНДРОМЕ АЛЬПОРТА СОПРОВОЖДАЕТСЯ
рахитом
глухотой
хондродистрофией
заболеваниями
глаз
СИНДРОМ
АЛЬПОРТА — ЭТО наследственный
нефрит мембранопролиферативный
гломерулонефрит системное заболевание
с повреждением почек
В
ОСНОВЕ РАЗВИТИЯ СИНДРОМА АЛЬПОРТА ЛЕЖИТ
диффузное утолщение гломерулярной БМ
нарушение
синтеза гломерулярной БМ потеря
подоцитами малых отростков
ОСТРЫЙ
НЕКРОЗ КАНАЛЬЦЕВ – ЭТО ПОВРЕЖДЕНИЕ
обратимое
необратимое
Установить
соответствие:
-
72.ОСТРЫЙ
НЕКРОЗПРИЧИНА
РАЗВИТИЯКАНАЛЬЦЕВ
1)
ишемический
А)
краш-синдром2)
пигментный
Б)
шок различной этиологии3)
токсический
В)
действие вирусных частицГ)
гемолитико-уремический синдром
Д)
действие солей тяжелых металлов
Ответ:
1-б
, 2-аг
, 3-д
.
ОСТРЫЙ
НЕКРОЗ КАНАЛЬЦЕВ ПОЧКИ, СВЯЗАННЫЙ С
ДЕЙСТВИЕМ ПИГМЕНТОВ,
РАЗВИВАЕТСЯ
ПРИ гемохроматозе краш-синдроме
гемолитических кризах шоке
различной этиологии
ТОКСИЧЕСКИЙ
ОСТРЫЙ НЕКРОЗ КАНАЛЬЦЕВ
ПОЧКИ
МОЖЕТ БЫТЬ СВЯЗАН С ДЕЙСТВИЕМ солей
ртути солей
кальция этиленгликоля
четыреххлористого
углерода
СТАДИИ
ОСТРОГО НЕКРОЗА КАНАЛЬЦЕВ ПОЧКИ основная
шоковая
олиганурическая
восстановления
диуреза нефротическая
ХАРАКТЕРИСГИКА
ОСНОВНОЙ СТАДИИ
ОСТРОГО
НЕКРОЗА КАНАЛЬЦЕВ ПОЧКИ устойчивая
олигурия выраженная
протеинурия
рецидивирующая
микро-, макрогематурия
ХАРАКТЕРИСГИКА
ОСТРОГО ТУБУЛОИНТЕРСТИ-ЦИАЛЬНОГО
НЕФРИТА
-
1)
атрофия
канальцев4)
фокальный
некроз канальцев2)
интерстициальный
отек5)
лейкоцитарная
инфильтрация3)
интерстициальный
фиброз6)
мононуклеарная
инфильтрация
ХАРАКТЕРИСГИКА
ОСТРОГО ИШЕМИЧЕСКОГО
НЕКРОЗА
КАНАЛЬЦЕВ ПОЧКИ фокальный некроз
канальцев диффузный
некроз канальцев фокальная
атрофия канальцев
окклюзия
просветов канальцев цилиндрами разрывы
базальной мембраны (тубулорексис)
В
РАЗВИТИИ ОСТРОГО ПИЕЛОНЕФРИТА ИГРАЮТ
РОЛЬ вирусная инфекция системные
заболевания бактериальная
инфекция обструкция мочевых путей
везикоуретеральный рефлюкс
НАИБОЛЕЕ
РАСПРОСТРАНЕННОЙ ПРИЧИНОЙ ПИЕЛОНЕФРИТА
ЯВЛЯЕТСЯ
ИНФЕКЦИЯ
восходящая
гематогенная
ОСНОВНЫЕ
ОСЛОЖНЕНИЯ ОСТРОГО ПИЕЛОНЕФРИТА
пионефроз
папиллярный некроз острый
некроз канальцев
паранефритический
абсцесс пилефлебитические абсцессы
печени
82.
ФОРМЫ ТУБУЛОИНТЕРСТИЦИАЛЬНОГО НЕФРИТА
обструктивный ксантогранулематозный
рефлюксная нефропатия анальгетическая
нефропатия лекарственный интерстициальный
ВАРИАНТЫ
УРАТНОЙ НЕФРОПАТИИ нефролитиаз
гидронефроз
острая
мочекислая нефропатия хроническая
подагрическая нефропатия
гемолитико-уремический
синдром взрослых
МАКРОСКОПИЧЕСКАЯ
ХАРАКТЕРИСТИКА ГИДРОНЕФРОЗА увеличенные
размеры отложения жира около лоханок
выраженная атрофия паренхимы
резко
растянутые чашечки и лоханки мелкозернистая
кортикальная поверхность незначительно
истонченное корковое вещество нормальные
или слегка уменьшенные размеры
ПРИ
ДОБРОКАЧЕСТВЕННОМ НЕФРОСКЛЕРОЗЕ
В
КРОВЕНОСНЫХ СОСУДАХ МОЖНО ОБНАРУЖИТЬ
гиалиноз
казеозный
некроз
фибриноидный
некроз луковичный артериолит продуктивный
васкулит концентрический артериолит
НАИБОЛЕЕ
ЧАСТЫЕ ИСТОЧНИКИ ЭМБОЛИЙ
ПОЧЕЧНЫХ
АРТЕРИЙ тромбы
глубоких вен голеней
тромбы
в аневризмах грудного отдела аорты
пристеночные
тромбы левого предсердия и желудочка
МАКРОСКОПИЧЕСКАЯ
ХАРАКТЕРИСТИКА ИНФАРКТА ПОЧКИ красный
клиновидный
неправильной
формы
белый с геморрагическим венчиком
основание обращено к поверхности почки
|
88. |
ПРИЧИНЫ |
|||
|
НА |
||||
|
1) |
камни |
4) |
стриктуры |
|
|
2) |
тромбы |
5) |
воспаление |
|
|
3) |
опухоли |
6) |
гиперплазия |
|
|
89. |
ОСНОВНЫЕ |
|||
|
1) |
4) |
цистиновые |
||
|
2) |
фосфаты |
5) |
пигментные |
|
|
3) |
оксалаты |
6) |
холестериновые |
СИНОНИМЫ
ПОЧЕЧНО-КЛЕТОЧНОЙ КАРЦИНОМЫ карциноид
гипернефрома
аденокарцинома
гипернефроидный
рак кортикальная
аденома
МАКРОСКОПИЧЕСКАЯ
ХАРАКТЕРИСТИКА ПОЧЕЧНО-КЛЕТОЧНОЙ
КАРЦИНОМЫ четкие
границы сферическая форма неправильная
форма желто-серо-белый
цвет обычно
бывает двусторонней
часто
прорастает в почечные вены обычно
располагается в области верхнего полюса
почки
КЛЕТКИ
ПАРЕНХИМЫ ПОЧЕЧНО-КЛЕТОЧНОГО РАКА ИМЕЮТ
ОБИЛЬНУЮ
СВЕТЛУЮ
ЦИТОПЛАЗМУ ЗА СЧЕТ НАКОПЛЕНИЯ липидов
гликогена
гранул
белковой природы вакуолей с изотонической
жидкостью
ОБСТРУКТИВНЫЕ
ЗАБОЛЕВАНИЯ МОЧЕТОЧНИКА МОГУТ ВЫЗВАТЬ
пиелонефрит
гидроуретер гидронефроз
везикоуретеральный
рефлюкс ретроперитониальный
фиброз
У
ЖЕНЩИН УРЕТРИТ ЧАСТО СОПРОВОЖДАЕТСЯ
-
1)
циститом
4)
простатитом
2)
проктитом
5)
парапроктитом
3)
аднекситом
6)
вульвовагинитом
НЕФРОТИЧЕСКИЙ
СИНДРОМ МОЖЕТ СОПРОВОЖДАТЬСЯ
геморрагическим синдромом тромботическими
и тромбоэмболическими осложнениями
ПРОЛИФЕРАЦИЯ
КЛЕТОК И ЛЕЙКОЦИТАРНАЯ ИНФИЛЬТРАЦИЯ
ПРИ ОСТРОМ
ПОСТСТРЕПТОКОККОВОМ
ГЛОМЕРУЛОНЕФРИТЕ ИМЕЮТ ХАРАКТЕР
фокальный диффузный
сегментарный
ПИЕЛОНЕФРИТ
ХАРАКТЕРИЗУЕТСЯ ВОСПАЛЕНИЕМ
канальцев
интерстиция
почечных лоханок почечных
клубочков
МЕХАНИЗМ
ОБРАЗОВАНИЯ ДЕПОЗИТОВ ПРИ
АНТИ-ГБМ-НЕФРИТЕ
иммунные
комплексы in situ
циркулирующие
иммунные комплексы
МЕХАНИЗМ
ОБРАЗОВАНИЯ ДЕПОЗИТОВ ПРИ
ОСТРОМ
СЫВОРОТОЧНОМ НЕФРИТЕ иммунные комплексы
in situ циркулирующие
иммунные комплексы
ГИСТОЛОГИЧЕСКАЯ
ХАРАКТЕРИСТИКА ОСТРОГО ВОСПАЛЕНИЯ
ПОЧЕЧНОГО
КЛУБОЧКА
отек стромы
пролиферация
мезангиоцитов склероз
и гиалиноз базальной мембраны
исчезновение
ножек отростков подоцитов инфильтрация
полиморфноядерными лейкоцитами
БЫСТРО
ПРОГРЕССИРУЮЩИЙ ГЛОМЕРУЛОНЕФРИТ ПРИ
СКВ ПО МЕХАНИЗМУ
РАЗВИТИЯ
антительный
иммунокомплексный
БЫСТРО
ПРОГРЕССИРУЮЩИЙ ГЛОМЕРУЛОНЕФРИТ
СОПРОВОЖДАЕТСЯ удвоением ГБМ формированием
полулуний
развитием
гиперклеточности клубочков
103.
Нефротический синдром у взрослых обычно
развивается при системных
заболеваниях первичном поражении почек
104.УДВОЕНИЕ
ГЛОМЕРУЛЯРНЫХ БАЗАЛЬНЫХ МЕМБРАН ПРИ
МЕМБРАНОПРОЛИФЕРАТИВНОМ
ГЛОМЕРУЛОНЕФРИТЕ ВЫЯВЛЯЮТ ПРИ ОКРАСКЕ
-
1)
суданом
III5)
пикрофуксином
2)
фукселином
6)
толуидиновым
синим3)
серебрением
7)
шифф-иодной
кислотой4)
конго-красным
гематоксилином
и эозином
ХРОНИЧЕСКИЙ
ГЛОМЕРУЛОНЕФРИТ ПРИ БЫСТРОПРОГРЕССИРУЮЩЕМ
ГЛОМЕРУЛОНЕФРИТЕ
РАЗВИВАЕТСЯ редко
часто
ПРИ
АМИЛОИДОЗЕ ПОЧЕК СМЕРТЬ БОЛЬНОГО МОЖЕТ
НАСТУПИТЬ ОТ ПОЧЕЧНОЙ
НЕДОСТАТОЧНОСТИ
острой
подострой
хронической
ПРИ
МЕМБРАНОЗНОЙ НЕФРОПАТИИ СМЕРТЬ БОЛЬНОГО
МОЖЕТ НАСТУПИТЬ ОТ
ПОЧЕЧНОЙ
НЕДОСТАТОЧНОСТИ острой подострой
хронической
ЛОБУЛЯРНЫЙ
ГЛОМЕРУЛОНЕФРИТ — ЭТО
наследственный
нефрит мембранопролиферативный
гломерулонефрит системное
заболевание с повреждением почек
СИСТЕМНАЯ
КРАСНАЯ ВОЛЧАНКА — ЭТО наследственный
нефрит первичный мембранопролиферативный
гломерулонефрит
системное
заболевание с повреждением почек 3
АМИЛОИДОЗ
ПОЧЕК – ЭТО ПРОЦЕСС обратимый необратимый
ХАРАКТЕРИСГИКА
ХРОНИЧЕСКОГО ТУБУЛОИНТЕРСТИ-ЦИАЛЬНОГО
НЕФРИТА
-
1)
атрофия
канальцев4)
фокальный
некроз канальцев2)
интерстициальный
отек5)
лейкоцитарная
инфильтрация3)
интерстициальный
фиброз6)
мононуклеарная
инфильтрация
112.
В РАЗВИТИИ ХРОНИЧЕСКОГО ПИЕЛОНЕФРИТА
ИГРАЮТ
РОЛЬ вирусная инфекция системные
заболевания бактериальная
инфекция обструкция мочевых путей
везикоуретеральный
рефлюкс
http://www.youtube.com/playlist?list=PLnbI8gbyR5iSrvoVMlul5S_j9AOfkVuro
ПРИ
ЗЛОКАЧЕСТВЕННОМ НЕФРОСКЛЕРОЗЕ В
КРОВЕНОСНЫХ СОСУДАХ МОЖНО
ОБНАРУЖИТЬ
гиалиноз
казеозный
некроз
фибриноидный
некроз луковичный
артериолит восковидный некроз
концентрический венулит
ПРИЧИНЫ
ОБСТРУКЦИИ МОЧЕВЫВОДЯЩИХ ПУТЕЙ
|
НА |
|||
|
1) |
камни |
4) |
стриктуры |
|
2) |
тромбы |
5) |
воспаление |
|
3) |
опухоли |
6) |
гиперплазия |
115.
ПРИЧИНЫ ОБСТРУКЦИИ МОЧЕВЫВОДЯЩИХ ПУТЕЙ
НА
УРОВНЕ МОЧЕВОГО ПУЗЫРЯ
-
1)
камни
4)
стриктуры
2)
тромбы
5)
воспаление
3)
опухоли
6)
гиперплазия
ткани соседнего органа
116.
ПРИЧИНЫ ОБСТРУКЦИИ МОЧЕВЫВОДЯЩИХ
ПУТЕЙ
|
НА |
|||
|
1) |
камни |
4) |
стриктуры |
|
2) |
тромбы |
5) |
воспаление |
|
3) |
опухоли |
6) |
гиперплазия |
|
117. |
ПРИЧИНЫ |
||||
|
НА |
|||||
|
1) |
камни |
4) |
стриктуры |
||
|
2) |
тромбы |
5) |
воспаление |
||
|
3) |
опухоли |
6) |
гиперплазия |
||
|
118. |
У |
||||
|
1) |
циститом |
4) |
простатитом |
||
|
2) |
проктитом |
5) |
парапроктитом |
||
|
3) |
аднекситом |
Нефротический синдром
Утратил силу — Архив
![]()
Версия: Клинические протоколы МЗ РК — 2014 (Казахстан)
Категории МКБ:
Нефротический синдром (N04)
Разделы медицины:
Нефрология
Общая информация
Краткое описание
Экспертным советом
РГП на ПХВ «Республиканский центр
развития здравоохранения»
Министерства здравоохранения
и социального развития
Республики Казахстан
от «12» декабря 2014 года
протокол № 9
Нефротический синдром – клинический синдром, характеризующийся тяжелой протеинурией >3,5г/1,73м2/сут, гипоальбуминемией, гиперлипидемией и отеками [1].
Тяжелая протеинурия всегда сопряжена с поражением почек при первичных ее заболеваниях, а также при широком круге системных и других заболеваний.
Так как понятие «синдром» еще не является диагнозом, то каждый случай нефротического синдрома требует проведения тщательного поиска возможных его причин, верификации морфологического варианта поражения почек и установления морфологического/клинического диагноза с последующим обоснованным лечения уже доказанного диагноза, а не синдрома.
I. ВВОДНАЯ ЧАСТЬ:
Название протокола: Нефротический синдром
Код протокола:
N04 – Нефротический синдром
N04.0 – Нефротический синдром, незначительные гломерулярные нарушения
N04.1 – Нефротический синдром, очаговые и сегментарные гломерулярные повреждения
N04.2 – Нефротический синдром, диффузный мембранозный гломерулонефрит
N04.3 – Нефротический синдром, диффузный мезангиальный пролиферативный гломерулонефрит
N04.4 – Нефротический синдром, диффузный эндокапиллярный пролиферативный гломерулонефрит
N04.5 – Нефротический синдром, диффузный мезангиокапиллярный (мембранопролиферативный) гломерулонефрит
N04.6 – Нефротический синдром, болезнь плотного осадка
N04.7 – Нефротический синдром, диффузный серповидный гломерулонефрит
N04.8 – Нефротический синдром, другие изменения
N04.9 – Нефротический синдром, неуточненное изменение
N08.0* Гломерулярные поражения при инфекционных и паразитарных болезнях, классифицированных в других рубриках
Гломерулярные поражения при: . малярии, вызванной Plazmodium malariae (B52.0+) . эпидемическом паротите (B26.8+) . шистосомозе [бильгарцилзе] (B65.-+) . септицемии (A40-A41+) . стронгилоидозе (B78.-+) . сифилисе (A52.7+)
N08.1* Гломерулярные поражения при новообразованиях
Гломерулярные поражения при: . множественной миеломе (C90.0+) .
макроглобулинемии Вальденстрема (C88.0+)
N08.2* Гломерулярные поражения при болезнях крови и иммунных нарушениях
Гломерулярные поражения при: . криоглобулинемии (D89.1+) . диссеминированном внутрисосудистом свертывании [синдроме дефибринации] (D65+) . гемолитико-уремическом синдроме (D59.3+) . пурпуре Геноха[-Шенлейна] (D69.0+) . серповидно-клеточных нарушениях (D57.-+)
N08.3* Гломерулярные поражения при сахарном диабете (E10-E14+ с общим четвертым знаком .2)
N08.4* Гломерулярные поражения при других болезнях эндокринной системы, расстройствах питания и нарушениях обмена веществ
Гломерулярные поражения при: . амилоидозе (E85.-+) . болезни Фабри(Андерсона) (E75.2+) . недостаточности лецитинхолестеринацилтрансферазы (E78.6+)
N08.5* Гломерулярные поражения при системных болезнях соединительной ткани
Гломерулярные нарушения при: синдроме Гудпасчера (M31.0+) . узелковом полиартериите (M30.0+) . системной красной волчанке (M32.1+) . тромботической тромбоцитопенической пурпуре (M31.1+) . гранулематозе Вегенера (M31.3+)
N08.8* Гломерулярные поражения при других болезнях, классифицированных в других рубриках
Гломерулярные нарушения при подостром бактериальном эндокардите (I33.0+)
Дата разработки протокола: 2014 год.
Сокращения, используемые в протоколе:
АГ – артериальная гипертензия
АНА – антинуклеарные антитела
АНЦА – антинейтрофильные цитоплазматические антитела
БКК – блокаторы кальциевых каналов
БМИ – болезнь минимальных изменений
БРА – блокаторы рецепторов ангиотензина
иАПФ – ингибиторы ангиотензинпревращающего фермента
ИГХ – иммуногистохимия
ИКН – ингибиторы кальцинейрина
ИФ – иммунофлюоресценция
МК – микофеноловая кислота
МКБ – Международная классификация болезней
ММФ – мофетила микофенолат
МН – мембранозная нефропатия
МНО – международное нормализованное отношение
МПГН – мембранопролиферативный гломерулонефрит
НМГ – низкомолекулярные гепарины
НС – нефротический синдром
ОПП – острое почечное повреждение
ПМР – пузырно-мочеточниковый рефлюкс
ПТИ – протромбиновый индекс
РКИ – рандомизированные клинические исследования
СД – сахарный диабет
СОЭ – скорость оседания эритроцитов
УЗИ – ультразвуковое исследование
ФСГС – фокально-сегментарных гломерулосклероз
ХБП – Хроническая болезнь почек
ХСН – хроническая сердечная недостаточность
ЦФ – циклофосфамид
ЭКГ – электрокардиограмма
ЭМ – электронная микроскопия
Категория пациентов: взрослые, беременные.
Пользователь протокола: нефрологи, терапевты, врачи общей практики, эндокринологи, ревматологи, гематологи, онкологи, акушер-гинекологи, врачи скорой помощи, фельдшеры.
Облачная МИС «МедЭлемент»
Облачная МИС «МедЭлемент»

Автоматизация клиники: быстро и недорого!
- Подключено 500 клиник из 4 стран
- 1 место — 800 RUB / 5500 KZT / 27 BYN в месяц
Классификация
Классификация нефротического синдрома:
I. По этиологическому фактору:
Первичные гломерулярные болезни (первичный идиопатический нефротический синдром):
• болезнь минимальных изменений;
• мембранозная гломерулопатия;
• фокально-сегментарный гломерулосклероз;
• мембрано-пролиферативный (мезангиокапиллярный) гломерулонефрит;
• другие пролиферативные гломерулонефриты.
Вторичные гломерулопатии (вторичный нефротический синдром) в рамках:
• инфекций: инфекционный эндокардит, сифилис, лепра, гепатиты В и С, мононуклеоз, цитомегаловирусная инфекция, ветряная оспа, ВИЧ, малярия, токсоплазмоз, шистосомиаз;
• применения медикаментов и наркотических средств: препараты золота, пеницилламин, НПВП, препараты висмута, лития, пробенецид, высокие дозы каптоприла, параметадон, героин;
• системных заболеваний: СКВ, синдром Шарпа, ревматоидный артрит, дерматомиозит, пурпура Шенлейн-Геноха, первичный и вторичный амилоидоз, полиартериит, синдром Такаясу, синдром Гудпасчера, герпетиформный дерматит, синдром Шегрена, саркоидоз, криоглобулинемия, язвенный колит;
• нарушения обмена веществ: сахарный диабет, гипотиреоз, семейная средиземноморская лихорадка;
• злокачественных новообразований: болезнь Ходжкина, неходжкинская лимфома, хронический лимфолейкоз, множественная миелома, злокачественная меланома, карциномы легких, желудка, толстой кишки, молочных желез, щитовидной железы, шейки матки, яичников и почек;
• аллергических реакций: укус насекомых, поллиноз, сывороточная болезнь;
• врожденные заболевания: синдром Альпорта, болезнь Фабри, нэил-пателла-синдром (синдром поражения ногтей и надколенника), серповидно-клеточная анемия, дефицит альфа1-антитрипсина;
• другие: преэклампсия, ПМР, IgА-нефропатия, стеноз почечной артерии (редко).
II. Морфологическая: по МКБ
• нефротический синдром, незначительные гломерулярные нарушения;
• нефротический синдром, очаговые и сегментарные гломерулярные повреждения;
• нефротический синдром, диффузный мембранозный гломерулонефрит;
• нефротический синдром, диффузный мезангиальный пролиферативный гломерулонефрит;
• нефротический синдром, диффузный эндокапиллярный пролиферативный гломерулонефрит;
• нефротический синдром, диффузный мезангиокапиллярный (мембрано-пролиферативный) гломерулонефрит;
• нефротический синдром, болезнь плотного осадка;
• нефротический синдром, диффузный серповидный гломерулонефрит;
• амилоидоз почек.
III. По активности:
• полная ремиссия – протеинурия < 300мг/сут;
• частичная ремиссия — снижение протеинурии на 50% от исходного уровня или < 2,0 г/сут;
• рецидив – вновь возникшая протеинурия после полной ремиссии или нарастание после частичной ремиссии.
IV. По состоянию функции почек:
Определение состояния функции почек основано на двух показателях — скорости клубочковой фильтрации (СКФ) и признаках почечного повреждения. Под повреждением почек понимаются структурные и функциональные изменения почек, выявленные в анализах крови, мочи (альбуминурия, протеинурия или гематурия) или при визуальных обследованиях. В зависимости от их сочетания выделяют пять стадий хронической болезни почек (ХБП).
ХБП— повреждение почек (альбуминурия более 30мг/сут, гематурия) либо снижение их функции в течение 3 месяцев и более. Определение и классификация ХБП внедрены Национальным почечным фондом, National Kidney Foundation (NKF) и рабочей группой по улучшению исходов почечных заболеваний, Kidney Disease Outcomes Quality Initiative (KDOQI) в 2002, [1-3].
|
Стадии ХБП |
Описание | СКФ (мл/мин/1,73м2) |
| 1 | Повреждение почек с нормальной или ↑СКФ | ≥90 |
| 2 | Повреждение почек с легким ↓СКФ | 60 – 89 |
| 3 | Умеренное ↓СКФ | 30 – 59 |
| 4 | Тяжелое ↓СКФ | 15 – 29 |
| 5 | Почечная недостаточность | ≤15 (или диализ) |
Диагностика
II. МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
Перечень основных и дополнительных диагностических мероприятий
Основные (обязательные) диагностические обследования, проводимые на амбулаторном уровне:
• ОАК;
• ОАМ;
• Биохимический анализ крови (общий белок, альбумин, креатинин, мочевина, К, Na, Ca, холестерин, сахар в сыворотке крови);
• Расчет СКФ по формуле Кокрофта-Голта;
• ИФА на вирусный гепатит В и С;
• Анализ крови на ВИЧ;
• УЗИ органов брюшной полости и почек;
Дополнительные диагностические обследования, проводимые на амбулаторном уровне:
• суточная протеинурия или протеин/креатининовый, альбумин/креатининовый коэффициенты;
• иммунологические методы исследования сыворотки крови: АСЛ-О, комплементы С3, С4, С50; АNA, anti-ds-DNA, p-,c-ANCA, С3-нефритический фактор, АТ к фосфолипидам, АТ к кардиолипину;
• определение концентрации уровня Циклоспорина-А в сыворотке крови: С0 (через 12ч от вечернего приема) и С2 (через 2 часа от утреннего приема);
• КТ грудного, брюшного сегментов.
Минимальный перечень обследования, который необходимо провести при направлении на плановую госпитализацию:
• ОАК;
• ОАМ;
• Биохимический анализ крови: (общий белок, альбумин, креатинин, мочевина, К, Na, Ca, холестерин, сахар в сыворотке крови);
• суточная потеря белка в моче;
• коагулограмма.
Основные (обязательные) диагностические обследования, проводимые на стационарном уровне:
• биопсия почки;
• ОАК: (до и после биопсии почки);
• ОАМ: (до и после биопсии почки);
• суточная протеинурия или протеин/креатининовый, альбумин/креатининовый коэффициенты;
• УЗИ почек (после биопсии);
• коагулограмма.
• морфологическая диагностика биоптата ткани почки:
световая микроскопия: гистохимия с окраской PAS, по Массону трихром, серебрение по Джонсу, Конго-красный на амилоид;
иммуногистохимия: с набором реагентов первичных антител и вторичных антител меченных пероксидазой хрена (CD) или;
иммунофлюоресценция: с набором реагентов первичных антител меченных флюорохромом;
Дополнительные диагностические обследования, проводимые на стационарном уровне:
• электронная микроскопия биоптата ткани почки;
• криоглобулины в сыворотке крови;
• иммунологические методы исследования сыворотки крови: АСЛ-О, комплементы С3, С4, С50; АNA, anti-ds-DNA, p-,c-ANCA, С3-нефритический фактор, АТ к фосфолипидам, АТ к кардиолипину, анти-ГБМ-антитела, антитела к PLAR2;
• электрофорез белков мочи: альфа-2-макроглобулин, Ig-G, трансферрин, альбумин, альфа-1-микроглобулин, ретинол-связывающий протеин, бета-2-микроглобулин;
• иммунофиксация белков крови на парапротеины: М-градиент, IgG, IgA, IgM, каппа, ламбда;
• иммунофиксация белков мочи на парапротеины: Бенс-Джонса, IgG, IgA, IgM, каппа, ламбда;
• определение концентрации уровня Циклоспорина-А в сыворотке крови: С0 (через 12ч от вечернего приема) и С2 (через 2 часа от утреннего приема);
• стернальная пункция;
• колоноскопия;
• ФГДС;
• рентген плоских костей черепа;
• КТ, МРТ брюшного, грудного сегментов, органов малого таза;
• УЗИ органов брюшной полости;
• УЗДГ сосудов почек;
• аудиограмма;
• онкомаркеры: СА 15-3; СА 19-9; СА 72-4; СА 125; РЭА; АФП; Cyfra 21-1; NSE RackPack; белок S 100, ПСА;
• ПЦР на HBV, HCV, вирус Эпштейн-Барра, ЦМВ, Парвовирус-В19;
• посев материала (моча, спинномозговая жидкость, мокрота) на микобактерии туберкулеза;
• ИФА антитела на гельминты и паразиты;
• токсикологические исследования: висмут, литий, параметадон, героин в крови и моче.
Диагностические мероприятия, проводимые на этапе скорой неотложной помощи:
• Измерение АД.
Диагностические критерии [2, 3]
Жалобы: появление отеков, снижение диуреза, изменения в анализах мочи (белок).
Анамнез: спонтанное появление и исчезновение отеков, кожной сыпи, эритемы, артралгий, выпадение волос, стоматиты, синуситы, риниты с кровянистым отделяемым, солнечная инсоляция, частые пневмонии, бронхиальная астма, эпизоды макрогематурии на фоне фарингита и ОРВИ, эпизоды лихорадки неясной этиологии, потеря веса за короткий промежуток времени, не связанная с желанием пациента, наличие вирусного гепатита, сопутствующие заболевания:
ревматоидный артрит, хронические очаги инфекции (н-р: остеомиелит, бронхоэктатическая болезнь и др.). Прием препаратов золота, висмута, лития, НПВП, пробенецид, высокие дозы каптоприла, параметадон; употребление героина.
У женщин – привычные выкидыши в анамнезе.
Аллергические реакции на контрастные вещества, вакцины.
Физикальное обследование:
|
Признак |
Характеристика | Возможные причины |
| Отеки | мягкие, различной степени выраженности: от минимальных периферических до полостных и анасарки | Нефротический синдром |
| Кожная эритема | мигрирующая рожеподобная эритема без определенной локализации с абдоминальной болью | Нефротический криз |
| На лице в виде «бабочки» | СКВ | |
| Кожная сыпь | В зоне «декольте» | СКВ |
| Симметричная на верхних, нижних конечностях, туловище | Системные заболевания, васкулиты, инфекции | |
| Поражение слизистых оболочек | Энантемы, хейлит, стоматит | Системные заболевания |
| Суставной синдром | В виде артралгий без деформаций | Системные заболевания |
| Мелких суставов кистей рук, с деформацией | Ревматоидный артрит → амилоидоз почек | |
| Выделения из дыхательных путей | Кровянистые выделения из носа, мокрота с примесью крови | Системные васкулиты → БПГН |
| Нарушение кожной чувствительности | Снижение чувствительности кожи верхних и/или нижних конечностей | Сахарный диабет |
| Ортостатическая гипотония | Разница между АД сидя и стоя | Сахарный диабет |
| Увеличение лимфатических узлов | Увеличение лимфоузлов различной локализации | Онкологические заболевания → паранеопластические нефропатии |
Лабораторные исследования:
Главными лабораторными критериями собственно нефротического синдрома являются:
• в анализе суточной экскреции белка в моче: протеинурия более 3,5 г/сут;
• в биохимическом анализе крови: гипопротеинемия, гипоальбуминемия, гиперлипидемия.
Наряду с восполнением критериев НС изменения следующих лабораторных показателей помогут в поиске причин вторичного НС.
|
Исследования |
Показатели | Возможные причины НС |
| Моча на суточную экскрецию белка | Более 3,5 г/сут | НС, рецидив НС |
| Микроскопия осадка мочи |
Дисморфные эритроциты эритроцитарные цилиндры |
+ нефритический синдром → БПГН |
| Общий анализ крови | Ускоренное СОЭ | Активность нефрита |
| Анемия |
Системные заболевания, Множественная миелома Онкологические заболевания |
|
| Лейкоцитоз |
Сепсис Инфекции |
|
| Лейкопения |
Системные заболевания Вирусные инфекции |
|
| Эозинофилия | Системные васкулиты (синдром Чарга-Стросса) → БПГН | |
| Тромбоцитопения | Системные заболевания | |
| Биохимические исследования крови |
↑ мочевины ↑ креатинина Изменения К+, Na+, Ca2+, PO43-, Cl—, HCO3— |
Снижение функции почек |
| Гипопротеинемия, гипоальбуминемия | Нефротический синдром, цирроз печени | |
| Гиперпротеинемия | Миеломная болезнь и др. парапротеинемии | |
| ↑ мочевая кислота | Синдром лизиса опухоли, Подагра | |
| ↑ ЛДГ | ГУС, гемолитические анемии | |
| ↑ Креатинкиназы | Травмы и метаболические болезни | |
| Биохимический анализ мочи | Na+, креатинин в моче для расчета экскретируемой фракции Na (FENa) | Преренальное и ренальное ОПП |
| Специфические иммунологические исследования крови | ↑ АНА, антитела к двуспиральной ДНК | СКВ |
| ↑ р- АНЦА | Системные васкулиты: микроскопический полиангиит | |
| ↑ с-АНЦА | Системные васкулиты: гранулематоз Вегенера, синдром Чарга-Стросса | |
| наличие анти-ГБМ-антител | Анти-ГБМ-нефрит (синдром Гудпасчера) | |
| ↑ титр АСЛ-О | Постстрептококковый ГН | |
| наличие Криоглобулинов | Криоглобулинемия (эссенциальная или при разных заболеваниях) | |
| наличие Антифосфолипидных антител (антикардиолипиновые антитела, волчаночный антикоагулянт) | АФС-синдром | |
| ↓ С3, ↓С4, ↓ СН50 | СКВ, инфекционный эндокардит, шунт-нефрит | |
| ↓ С3, ↓ СН50 | Постстрептококковый ГН | |
| ↓С4, ↓ СН50 | Эссенциальная смешанная криоглобулинемия | |
| ↓ С3, ↓ СН50 | С3-гломерулопатия | |
| ↑ Прокальцитонина | Сепсис | |
| ↑ анти-PHLAR2 | Идиопатическая мембранозная нефропатия | |
| Иммунофиксация белков крови и мочи |
↑ легкие (каппа, лямбда), тяжелые цепи иммуноглобулинов ↑ парапротеины Бенс-Джонса |
Плазмаклеточные дискразии, миеломная болезнь, амилоидоз |
| Онкомаркеры | ↑ СА 15-3; СА 19-9; СА 72-4; СА 125; РЭА; АФП; Cyfra 21-1; NSE RackPack; белок S 100, ПСА; | Паранеопластическая нефропатия |
Инструментальные исследования:
|
Исследования |
Характерные изменения | Возможные причины НС |
| УЗИ органов брюшной полости и почек |
Асцит Увеличение размеров почек |
НС, рецидив НС |
| Морфологическое исследование биоптата почечной ткани |
Отсутствие изменений при световой микроскопии Отрицательная иммуногистохимия (ИГХ) и иммунофлюоресценция (ИФ) Сглаживание ножек подоцитов при электронной микроскопии (ЭМ) |
Болезнь минимальных изменений |
|
Склероз отдельных сегментов (сегментарность) в части (фокальность) клубочков Расширение мезангиального матрикса Эндокапиллярная гиперклеточность и гиалиноз Отрицательная ИФ/ИГХ или наличие IgM, C3 При ЭМ сглаживание ножек и дегенерация подоцитов |
Фокально-сегментарный гломерулосклероз (клеточный, верхушечный, коллабирующий, перихилярный варианты) | |
|
При световой микроскопии: отсутствие изменений на ранних стадиях и диффузное утолщение ГБМ в поздних стадиях Наличие отложений в виде «шипиков» на наружной поверхности (субэпителиально) ГБМ При ИГХ/ИФ: гранулярное свечение IgG ± C3 депозитов в толще капиллярных стенок, IgG 4 субкласс При ЭМ: субэпителиальные депозиты |
Мембранозная нефропатия | |
|
При световой микроскопии: мезангиальная гиперклеточность, наличие иммунных депозитов в мезангии и субэпителиальном пространстве ИГХ/ИФ: гранулярное свечение IgG и С3 депозитов в капиллярах и мезангии |
Мезангиокапиллярный (мембрано-пролиферативный) гломерулонефрит, тип I | |
|
непрерывные, плотные, лентовидные депозиты в толще ГБМ Наличие С3 без иммуноглобулинов при ИФ/ИГХ |
Болезнь плотных депозитов (по старой классификации МПГН, тип II) | |
| Наличие иммунных депозитов аналогичных как и при МПГН, тип 1, но c полным нарушением ГБМ и мемебранозными изменениями | Мезангиокапиллярный (мембрано-пролиферативный) гломерулонефрит, тип III | |
| Изолированное присутствие С3 (+ снижение в крови уровня комплемента С3) | С3-гломерулопатия (выделен в отдельную форму из группы МПГН) | |
| Гиперклеточность и наличие полулуний в Боуменовом пространстве в > 50% клубочков | Экстракапиллярный гломерулонефрит (БПГН) | |
|
При окраске Конго красным наличие аморфных экстрацеллюлярных амилоидных депозитов в мезангии, ГБМ и кровеносных сосудах, и реже присутствие их в канальцах и интерстиции ИГХ/ИФ: свечение лямбда, иногда каппа ЭМ: наличие фибриллов |
Амилоидоз почки |
Показания для консультации специалистов:
• консультация онколога (при подозрении на паранеопластическую нефропатию);
• консультация фтизиатра (при подозрении на спец.процесс);
• консультация ревматолога (при подозрении на системное заболевание);
• консультация окулиста (для осмотра глазного дна);
• консультация хирурга (при нефротических кризах для исключения острой хирургической патологии);
• консультация гепатолога (при HBV-, HCV-ассоциированных гломерулонефритах);
• консультация невролога (при подозрении на нейролюпус, системный васкулит);
• консультация гематолога (при подозрении на миеломную болезнь, болезнь легких цепей);
• инфекциониста – при длительной лихорадке, инфекциях.
Дифференциальный диагноз
Дифференциальный диагноз
Дифференциации требуют заболевания, которые протекают с выраженным отечным синдром: НС, цирроз печени, застойная хроническая сердечная недостаточность (ХСН).
|
Признак |
Нефротический синдром | Цирроз печени | Застойная ХСН |
| Начало заболевания |
С появления отеков на лице, на ногах (дни, недели). Может принимать волнообразное течение с эпизодами спонтанного схождения отеков |
С постепенного появления асцита | Постепенное нарастание отеков на нижних конечностях (месяцы, годы) |
| Жалобы | Появление отеков | Увеличение объема живота, кровоточивость десен | Одышка, утомляемость |
| Анамнез |
Может быть связь с переохлаждением, приемом НПВП, героина, пеницилламина, препаратов золота, висмута, вакцинацией. При вторичном НС – системные заболевания |
Наличие ВГ-В, С, злоупотребление алкоголем | Длительная АГ, ИБС, перенесенный ИМ, ОНМК |
| Отеки | Мягкие, от минимальных до анасарки, легко перемещающиеся в зависимости от положения тела | Преимущественно асцит | Плотные, на нижних конечностях с трофическими изменения кожи |
| Артериальное давление | N (50%), иногда гипотония | Нормальное или склонность к гипотонии | Чаще повышенное, реже гипотония |
| Наследственность | Не отягощена | Не отягощена | Отягощена по АГ, СД, ожирению. Случаи внезапной смерти в семье |
| Протеинурия | Более 3,5 г/сут | Не характерна или минимальная | от минимальной до нефротического уровня |
| Гиперазотемия | Транзиторная на фоне активности НС, нарастает в зависимости от срока давности болезни | При развитии гепато-ренального синдрома, чаще при преренальной ОПП | Зависит от срока давности болезни и момента диагностики. Может быть исходная ХБП (гипертоническая / диабетическая нефропатия) |
| Биохимический анализ крови | Гипопротеинемия, гипоальбуминемия, гиперхолестеринемия | Гипопротеинемия, гипоальбуминемия, Возможны Гиперферментемия (АЛТ, АСТ, ГГТП), гипербилирубинемия | Возможна гиперхолестеринемия |
| Общий анализ крови | Ускоренное СОЭ, при аутоиммунных заболеваниях может быть анемия, тромбоцитопения, лейкопения | Тромбоцитопения | Сгущение крови: повышение гематокрита, снижение СОЭ |
| Коагулограмма | Может быть склонность к гиперкоагуляции | Гипокоагуляция (снижение ПТИ) | Не изменена или склонность к гиперкоагуляции |
| ЭхоКГ | Не характерны изменения | Не характерны изменения | Снижение ФВ <60%, диастолическая дисфункция, признаки ГЛЖ |
| ЭКГ | Не характерны изменения | Не характерны изменения | Признаки ГЛЖ |
Лечение
Цель лечения:
• достижение полной или частичной ремиссии;
• ликвидация экстраренальных симптомов (АГ, отеки) и осложнений (электролитные нарушения, инфекции, нефротический криз);
• замедление прогрессирования ХБП и отдаление ее терминальной стадии (диализ, трансплантация почки).
Немедикаментозное лечение:
Режим:
• свободный
• постельный при тяжелом состоянии пациента и наличии осложнений
• дозированная физическая активность по 30 минут 5 раз в неделю.
• отказ от курения;
• отказ от алкоголя.
Диета:
• сбалансированная, адекватное введение белка (1,5-2г/кг), калораж по возрасту;
• при наличии отеков и АГ – ограничение употребления натрия хлорид (поваренной соли) < 1-2г/сут;
Стационарно проводится диагностическая биопсия и установление морфологического диагноза и начало патогенетической терапии, которую следует продолжить в амбулаторных условиях.
Медикаментозное лечение:
Лечение отеков. Диуретики назначают при значительных отеках (назначение кортикостероидов обычно приводит к восстановлению диуреза на 5-10 день). Диуретики не назначают при рвоте, диарее, гиповолемии.
При длительно сохраняющихся отеках назначают Фуросемид 1-3мг/кг/сут внутривенно однократно болюсно или Торасемид 5-10-20мг внутрь. Для пациентов с рефрактерными отеками используется комбинация петлевых и тиазидоподобных (Индапамид 2,5-5мг) диуретиков и/или калий-сберегающих диуретиков (Спиронолактон 25-100мг/сут), в тяжелых случаях (риск развития нефротического криза) — комбинация диуретиков и альбумина (10%-100мл) [17].
При резистентных отеках в сочетании с почечной недостаточностью острого периода показано проведение гемодиализа с ультрафильтрацией до выхода из критического состояния почечной недостаточности и появления диуреза.
Лечение артериальной гипертензии. При отсутствии симптомов острого почечного повреждения назначаются с антигипертензивной целью ингибиторы АПФ (Периндоприл 5-20мг/сут) или БРА (Вальсартан 80-320мг/сут), но не в комбинации. При сохраняющейся гипертонии или наличии ОПП назначают блокаторы Са-каналов (Амлодипин 5-30мг/сут, Дилтиазем 90-320мг/сут, Нифедипин 10-30мг сублингвально как средство скорой помощи). При наличии тахикардии и/или симптомов ХСН дополнительно назначают бета-блокаторы – карведилол 12,5-50мг/сут в 2 приема внутрь.
Патогенетическая терапия НС отличается в зависимости от морфологического варианта, поэтому правомочна только после проведения биопсии почки, верификации морфологического диагноза и должна начинаться в стационаре и продолжиться в амбулаторных условиях.
Терапия болезни минимальных изменений (Minimal change disease). Данный морфологический вариант (гломерулонефрит с минимальными изменениями – ГНМИ) встречается в 10-20% биопсии при НС у взрослых.
При установлении причины болезни минимальных изменений требуется проведение этиотропного лечения заболевания, приведшего к развитию НС:
• обнаружение паразитов, гельминтов – антипаразитарное/противогельминтное лечение;
• новообразования – химиотерапия, лучевая терапия, оперативное вмешательство;
• диабетическая нефропатия – лечение СД и нефропротективная терапия;
Лечение дебюта. Метилпреднизолон 0,6-0,8мг/кг/сут или преднизолон 0,5-1мг/кг/сут (максимальные суточные дозы 64 и 80мг, соответственно) в течение 4-6-8 недель (предпочтительнее длительное назначение до 12-16 недель) в виде однократного приема в утреннее время, после приема завтрака (Уровень 1В). По достижению полной или частичной ремиссии – снижение дозы преднизолона на 5 мг каждые 3-4 дня до достижения дозы преднизолона 20-30мг/сут. Последующие 2-3 месяца прием преднизолона в альтернирующем режиме, т.е. через день с постепенным снижением дозы по 5мг каждые 1-2 недели, до достижения 10мг (Уровень 2В). Последующее снижение дозы по 2,5мг каждые 1-2 недели при альтернирующей схеме приема до полной отмены [4-6].
Более быстрое снижение дозы преднизолона возможно при появлении нежелательных явлений и осложнений стероидной терапии.
Пациенты, не достигшие полной или частичной ремиссии после приема полной дозы в течение 16 недель определяются как стероид-резистентные и требуют комбинированной терапии циклоспорином-А и минимальной дозой преднизолона 0,15-0,2мг/кг/сут [5, 7-9].
Лечение рецидива. У 50-75% пациентов, ответивших на стероидную терапию встречаются рецидивы. При рецидиве назначается Преднизолон в дозе 60 максимум 80мг/сут в течение 4-х недель с последующим снижением по 5 мг каждые 3-5 дней до полной отмены в течение 1-2 мес [7, 8].
При частых (3 и более в течение 1 года) рецидивах или стероид-зависимой (рецидив на фоне приема стероидов) форме используется комбинированная терапия: низкие дозы преднизолона 0,15-0,20мг/кг/сут + один из следующих групп препарат: алкилирующие агенты (циклофосфамид), ингибиторы кальцинейрина (циклоспорин-А или Такролимус), антиметаболиты (микофеноловая кислота, мофетила микофенолат) [10].
Циклофосфамид 2мг/кг/сут, внутрь в течение 8-12 нед, под контролем числа лейкоцитов (не менее 3 х 109/л) + профилактика геморрагического цистита (месна, урометиксан) [10, 11].
Циклоспорин-А микроэмульсионная форма в дозе 3-5мг/кг/сут, внутрь в 2 приема, не микроэмульсионная форма в дозе 4-5мг/кг/сут, внутрь в 2 приема, при целевой С0 концентрации 100-200нг/мл в течение 18-24 месяцев и более (Уровень 2В). [8-11].
Микофеноловая кислота, внутрь в дозе 540-720мг х 2 раза или мофетила микофенолат внутрь в дозе 750-1000мг х 2 раза в день в течение 6-26 месяцев [8].
Ритуксимаб – химерическое моноклональное антитело, используется для лечения различных морфологических вариантов НС. Рекомендуется лишь при отсутствии эффекта от выше проведенной терапии. Назначается в дозе 375мг/м2 поверхности тела, 1 раз в неделю, всего №4, внутривенно, капельно после премедикации.
При стероид-резистентности рекомендуется проведение повторной биопсии (так как не исключается трансформация в ФСГС).
При персистирующем НС несмотря на проводимую иммуносупрессивную терапию, должны назначаться иАПФ или БРА, диуретики (петлевые + тиазидоподобные + антагонисты альдостерона), статины (Аторвасататин 10-40мг/сут), антикоагулянты (низкомолекулярные гепарины) для профилактики тробоэмболических осложнений.
У пациентов с ОПП при БМИ проводят заместительную почечную терапию (гемодиализ, гемодиафильстрацию, ультрафильтрацию при наличии показаний , но с одновременным применением кортикостероидов. (2D) Не рекомендуется применять статины для коррекции гипрелипидемии и не применять иАПФ или БРА у нормотензивных пациентов для снижения протеинурии при лечении первого эпизода нефротического синдрома при БМИ. (2D). При наличии БПГН с повышением уровня креатинина более 500мкмоль/л, обусловленного системными васкулитами и синдромом Гудпасчера показано проведение плазмафереза (непрерывным методом) через день в течение 2-х недель со 100% заменой плазмы. Замещение необходимо проводить альбумином 10%.
Лечение очаговых и сегментарных гломерулярных повреждений, ФСГС. (Focal Segmental Glomerulosclerosis, FSGS). ФСГС бывает первичным и вторичным. Поэтому необходимо внимательно собрать анамнез и провести дополнительные исследования для поиска / исключения ФСГС, ассоциированного с ВГ-В,С, с ВИЧ и другими состояниями (АГ, ожирение, героиновая зависимость).
При установлении ФСГС в иммуносупрессивной терапии не нуждаются (Уровень 2В):
— пациенты с диагностированной ФСГС, нормальной функцией почек и протеинурией менее 3,0г/сут (существует возможность спонтанной ремиссии);
— пациенты со сниженной функцией почек и с протеинурией менее 3,0г/сут (не исключается, что эти пациенты к моменту диагностики ФСГС имеют снижение уровня протеинурии по сравнению с предыдущим недиагностированным периодом) [12].
Пациентам без противопоказаний к стероидной терапии назначается Метилпреднизолон или Преднизолон (Уровень 1А) в дозе 1мг/кг/сут (максимум 80мг/сут), внутрь. Длительность приема и начало снижения дозы зависят от скорости достижения полной или частичной ремиссии. Пациентам с сопутствующими заболеваниями (тяжелый остеопороз, диабет, ожирение) инициальную терапию мы рекомендуем начинать с комбинации (Уровень 2В) низкой дозы Преднизолона (0,15-0,20мг/кг/сут, максимум 15мг/сут) + Циклоспорин (2-4мг/кг/сут, разделенных на 2 приема) или Такролимус (4мг/сут, внутрь, разделенных на 2 приема). Целевой уровень концентрации С0 циклоспорина в сыворотке крови 100-175нг/мл, такролимуса 4-7нг/мл. [13-15].
При СКФ < 40мл/мин применение ингибиторов кальцинейрина не рекомендуется по причине их нефротоксичности.
Для всех пациентов с ФСГС рекомендуется назначение нефропротективной терапии иАПФ или БРА (Уровень 1 В).
Пациентам с НС и ХБП рекомендуется назначение статинов (Уровень 2В). Пациентов, получающих терапию циклоспорином и статинами, необходимо мониторировать на предмет рабдомиолиза (при взаимодействии циклоспорина и статинов). [16-18].
При резистентном нефротическом синдроме используется Ритуксимаб в дозе 375мг/м2 поверхности тела, 1 раз в неделю, всего №4, внутривенно, капельно после премедикации или по 1000мг №2 внутривенно, капельно с интервалом в 2 недели.
Лечение диффузного мембранозного гломерулонефрита. Мембранозная нефропатия (МН) является наиболее частой причиной НС у взрослых и может выявляться в 30-50% биопсированных случаев. Причиной вторичной МН могут быть инфекции (ВГ-В,С, опухолевые заболевания, в частности неходжкинские лимфомы и др, лекарства, системные заболевания). Поэтому прежде, чем приступить к патогенетической терапии, необходимо провести дополнительные исследования для поиска / исключения выше названных причин вторичной МН. В течении МН может наблюдаться спонтанная полная ремиссия протеинурии в 5-30% случаев в течение 5 лет [18-19], частичная ремиссия – в 25-40% в течение 5 лет [16]. Терминальная стадия ХБП у не леченных пациентов отмечается в 14% в течение 5 лет, 35% в течение 10 лет и 41% в течение 15 лет [20].
Поэтому иммуносупрессивную терапию необходимо начинать лишь в случаях необъяснимых причин повышения креатинина сыворотки крови, суточной протеинурии >4г/сут.
Иммуносупрессивная терапия включает чередование кортикостероидов (преднизолон 0,5мг/кг/сут или метилпреднизолон 0,4мг/кг/сут) в месяцы 1, 3, 5, начиная с пульс-терапии в начале каждого месяца + циклофосфамид в дозе 2-2,5мг/кг/сут, внутрь, в месяцы 2, 4, 6. Альтернативой являются ингибиторы кальцинейрина (циклоспорин или такролимус, дозировки как и при ФСГС) в сочетании с минимальной дозой преднизолона (метилпреднизолона). ЦФ предпочтителен в случаях снижения СКФ < 30%, в других ситуациях предпочтительно начинать с ингибиторов кальцинейрина (циклоспорина-А, как наиболее изученного препарата при МН, по сравнению с такролимусом). Длительность лечения ИКН составляет 18-24 и более месяцев.
При отсутствии эффекта возможно применение ритуксимаба в аналогичной дозе и длительности, как и при ГНМИ и ФСГС.
В качестве не иммуносупрессивной терапии следует применять иАПФ, БРА, статины, диуретики (петлевые, тиазидоподобные, антагонисты альдостерона в маленькой дозе 12,5-25мг/сут), препараты для профилактики тромбоэмболических осложнений (НМГ в профилактической или терапевтической дозе).
Лечение диффузного мезангиального пролиферативного гломерулонефрита. Первая линия терапии включает нефропротективную терапию иАПФ или БРА, статины при повышении ЛПНП. Лечение Ig-A-нефропатии иммуносупрессантами проводится в случае протеинурии >1,0г/сут, повышения креатинина крови и морфологических признаках активности (пролиферативные и некротизирующие изменения клубочков) при биопсии [24-29].
Иммуносупрессивная терапия подразумевает монотерапию кортикостероидами, а при быстропрогрессирующем течении и находке полулуний при нефробиопсии требует проведения комбинированной иммуноспурессивной терапии. Кортикостероиды в дебюте в виде пульс-терапии 15мг/кг веса в/в капельно №3, затем внутрь в дозе 1мг/кг/сут 60 дней, затем 0,6мг/кг/сут 60 дней, затем 0,3мг/кг/сут 60дней + циклофосфамид 0,5мг/м2, в/в капельно ежемесячно в течение 6 месяцев. Во второй линии вместо циклофосфамида возможно применение микофеноловой кислоты в дозе 360-1440мг/сут в 2 приема или Мофетила микофенолата 500-2000мг/сут в 2 приема. (Не забывать тератогенное действие Микофенолата и микофеноловой кислоты и при планировании беременности отказаться от данной группы препаратов). [30-31].
Лечение диффузного мезангиокапиллярного гломерулонефрита (мембранопролиферативный гломерулонефрит – МПГН).
Лечение первичного заболевания (гепатиты В и С). При МПГН 1 типа используют длительное лечение преднизолоном в альтернирующем режиме (30-60 мг/м2 / 48 час). В некоторых исследованиях показана эффективность лечения Мофетила микофенолатом. [34-46].
Лечение гломерулярных поражений при новообразованиях – лечение проводится в специализированном онкологическом учреждении и направлено на устранение причины нефротического синдрома.
Лечение гломерулярных поражений при болезнях крови и иммунных нарушениях.
При смешанной криоглобулинемии, ассоциированной с вирусным гепатитом С, нефротической протеинурией и признаками прогрессирующего заболевания почек или с явным обострением криоглобулинемии предлагается лечение плазмаферезом, ритуксимабом в сочетании с метилпреднизолоном внутривенно и проведение сопутствующей противовирусной терапии (2D).
При ГУС – плазмаферез непрерывным методом, с замещением 100% плазмы, через день в течение 2-х недель.
При пурпуре Шенлейн-Геноха лечение проводится в соответствии с морфологическим вариантом нефротического синдрома (см. лечение морфологических форм).
Лечение гломерулярных поражений при сахарном диабете (диабетическая нефропатия с нефротическим синдромом).
Коррекция АД и нефропротективная терапия: ИАПФ или БРА (но не в комбинации) при альбуминурии 30-299мг/сут (Уровень С), при альбуминурии > 300мг/сут (Уровень А). АГ является одним из важнейших независимых факторов риска прогрессирования ДН/ХБП [4]. Целевой уровень АД при ДН/ХБП составляет ≤ 140/90мм.рт.ст., при наличии альбуминурии или протеинурии ≤ 130/80мм.рт.ст. Препаратами выбора в лечении артериальной гипертензии при ДН/ХБП являются иАПФ (периндоприл 5мг, периндоприл 5мг/индапамид 1,25мг, периндоприл 10мг/индапамид 2,5мг) и БРА (Валсартан 80, 160мг 2 раза в день; Эпросартан 600мг, Лозартан 50мг), т.к. помимо антигипертензивного эффекта они снижают протеинурию, снижают ренальную симпатическую активность и играют роль в ремоделировании сердца. Возможно использование диуретиков при наличии перегрузки жидкостью и натрием у пациентов без соль-теряющей нефропатии. Тиазидоподобные диуретики (Индапамид 1,25-2,5мг 1 раз в день) могут использоваться на ранних стадиях ХБП, но нужно помнить, что они не эффективны при СКФ ≤ 30мл/мин/1,73м2. В 4-5 стадии ХБП применимы петлевые диуретики (Торасемид 10-40мг 1 раз в день). Антигипертензивными препаратами следующей линии являются Бета-блокаторы (карведилол 12,5-50мг в 2 приема) и Блокаторы кальциевых каналов (Амлодипин 5-30мг/сут). Но необходимо помнить, что дигидропиридиновые БКК (нифедипин, амлодипин) повышают внутриклубочковое давление и усиливают протеинурию, что снижает их полезное действие в плане замедления прогрессирования ХБП по сравнению с другими группами антигипертензивных препаратов. Подбор дозы препарата должен проводиться с учетом СКФ [8].
При резистентном НС ДН необходимо провести биопсию почки и определить морфологический вариант возможного иммуного гломерулярного поражения почек и назначить лечение соответственно морфологическому варианту поражения почек (лечение в зависимости от морфологического варианта см выше).
Лечение гломерулярных поражений при системных болезнях соединительной ткани:
— рекомендуется начинать иммуносупрессию Циклофосфамидом 2мг/кг/сут в течение 3-х мес или 600-1000мг в/в капельно 1 раз в месяц, кортикостероидами, Метилпреднизолон 500-1000мг/сут в течение 3-х дней с переходом прием внутрь Преднизолон 1мг/кг веса (максимум 60мг/сут) с последующим снижением и отменой через 6 месяцев и Плазмаферезом.
При узелковом полиартериите, гранулематозе Вегенера, синдроме Чарга-Стросса
Циклофосфамид 600-800мг в/в капельно 1 раз в месяц в течение 6 месяцев, Метилпреднизолон 500-1000мг/сут в течение 3-х дней с переходом на прием внутрь Преднизолон 1мг/кг веса (максимум 60мг/сут) с последующим началом снижениия через 4-6 недель и до поддерживающей дозы 10-15мг/48ч.
Лицам фертильного возраста вместо Циклофосфамида предпочтительней использовать Ритуксимаб в дозе 375мг/м2, № 4 в/в капельно (после премедикации) еженедельно или по 1000мг в/в капельно (после премедикации) №2 с интервалом 2 недели.
– лечение зависит от класса волчаночного нефрита (ВН):
Класс I ВН (минимальный мезангиальный ВН) – лечение пациента проводится в зависимости от выраженности внепочечных проявлений.
Класс II ВН (мезангио-пролиферативный ВН) – при уровне протеинурии < 1г/сут лечение проводится в зависимости от наличия внепочечных проявлений, при уровне протеинурии > 1г/сут показана комбинированная терапия метилпреднизолона / преднизолона 0,5-1мк/кг веса и микофеноловой кислоты 360-720мг х 2 раза в день / мофетила микофенолат 500-1000мг х 2 раза в день в течение 2-х лет. При уровне протеинурии > 3г/сут лечениепроводится аналогично как и при БМИ – Преднизолон и/или Циклоспорин-А (ЦсА). Дозы, режимы и длительность такие же.
Класс III ВН (очаговый ВН) и класс IV ВН (диффузный ВН). Инициальная терапия заключается в назначении кортикостероидов (метилпреднизолон в виде пульс терапии по 500-1000мг в/в капельно в течение 3-х дней с переходом на прием внутрь преднизолон 0,5-1мг/кг веса,) в сочетании с Циклофосфамидом (ЦФ) (1В) или Микофеноловой кислотой (МК) 1440-2160мг/сут / Мофетилом микофенолатом (ММФ) 2000-3000мг/сут (1В) до 6мес. Для пациентов не ответивших на терапию ЦФ и ММФ назначается Ритуксимаб в дозе 1000мг в/в капельно (после премедикации) №2 с интервалом в недели. Белимумаб назначается при преобладании экстраренальных симптомов СКФ. Поддерживающая терапия после 6 месяцев инициальной терапии проводится МК 720-1440мг/сут / ММФ 1000-2000мг/сут и низкими дозами кортикостероидов внутрь (эквивалентными ≤10мг/сут преднизолона) (1В). Пациентам которые не переносят МК/ММФ рекомендуется использовать ЦсА. Если полная ремиссия не достигнута в течение 12 месяцев поддерживающей терапии, следует решить вопрос о повторной биопсии почки.
Класс V ВН (мембранозный ВН). Пациентам с нормальной функцией почек и субнефротической протеинурией назначается с антипротеинурической / антигипертензивной целью иАПФ / БРА. Кортикостероиды и иммуносупрессанты показаны если имеются показания со стороны внепочечных проявлений СКВ (2D). Лечение пациента с персистирующим НС проводится кортикостероидами в сочетании с иммуносупрессивными препаратами: ЦФ или ЦсА (2C) или МК/ММФ (2D). Дозы и режимы как и при других вариантах ВН.
Класс VI ВН (склерозирующий ВН) – кортикостероиды и иммуносупрессанты назначаются только лишь при наличии внепочечных проявлений СКВ.
Дополнительная терапия при СКВ включает:
— назначение иАПФ/БРА с достижением целевого уровня АД ≤130/80мм.рт.ст.
— лечение осложнений ХБП: анемии (эпоэтин-альфа 2000МЕ 2-3 раза в неделю п/к, эпоэтин бета, дарб-эпоэтин, метоксиполиэтиленгликоль эпоэтин бета), вторичного гиперпаратиреоидизма (альфакальцидол 25-50мкмг х 1 раз в день, мимпара 30-160мг х 1 раз в день).
— Гидрохлорохин 200-400мг внутрь 1 раз в день всем пациентам с СКВ при отсутствии противопоказаний.
Лечение гломеруляпных поражений при амилоидозе.
продолжить лечение основного заболевания (хронические инфекции, хронические воспалительные заболевания, н-р ревматоидный артрит) для профилактики отложения новых амилоидных отложений.
Для лечения хронической инфекции использовать антибактериальную терапию и при показаниях хирургическое вмешательство.
Для лечения хронических воспалительных заболеваний показывают свою эффективность следующие группы агентов: алкилирующие агенты (ЦФ и хлорамбуцил), анти-ФНО терапия (Инфликсимаб, Этанерцепт, Адалимумаб), антитела к рецепторам ИЛ-6 (Тоцилизумаб). Колхицин эффективен при семейной форме амилоидоза.
АL-амилоидоз
: целью терапии является снижение или элиминация плазмаклеточных клонов. Режимы терапии включают ЦФ, Талидомид и Дексаметазон (СТД-схема) или Мелфалан + Дексаметазон (М/Декс-схема). Ответ на терапию наблюдается в 30-50% случаях. Альтернативой является применение Бортезомиба 1,3 мг/м2 в дни 1, 4, 8 и 11 (циклы 1, 3 и 5), дексаметазон 40 мг внутрь в дни 1-4, 9-12, 17-20 (циклы 2, 4 и 6). При отсутствии успеха используется высокодозная химиотерапия с трансплантацией аутологичных стволовых клеток. При наступлении терминальной стадии ХБП – диализ. Трансплантация почки возможна только при достижении полной ремиссии плазмаклеточной дискразии, иначе ожидается рецидив в трансплантате.
Лечение гломерулярных поражений при множественной миеломе:
при гиперкальциемии – регидратация в/в введением кристаллоидов > 5л, бисфосфонаты в/в показаны при сохраняющейся гиперкальцемии в эуволемическом состоянии;
при системном ацидозе – бикарбонат натрия 4%;
избегать назначения петлевых диуретиков (фуросемид и торасемид);
при гиперурикемии – аллопуринол 100мг в сутки;
при анемии – высокие дозы эритропоэтинов (Эпоэтин альфа, бета 40 000МЕ/нед п/к);
при почечной недостаточности – гемодиализ до выхода из состояния ОПП;
плазмаферез – для удаления циркулирующих легких цепей;
специфическая терапия: Бортезомиб 1,3 мг/м2 в дни 1, 4, 8 и 11 (циклы 1, 3 и 5), дексаметазон 40 мг внутрь в дни 1-4, 9-12, 17-20 (циклы 2, 4 и 6). При безуспешной консервативной терапии показана пересадка аутологичных стволовых клеток.
Другие медикаменты:
− Антациты (Алмагель по 1 д.л. х 3 раза в день) или блокаторы протонной помпы (Рабепразол 10-20мг х 1-2 раза в день; Омепразол 10-20мг х 1-2 раза в день) при появлении гастроинтестинальных симптомов.
− Карбонат кальция (250-500мг/сут) длительно необходим, если терапия преднизолоном продолжается более 3 месяцев [15].
− Витамин Д, 800МЕ/сут в течение 2-3 мес. под контролем уровня фосфора и ПТГ;
− Обычно нет необходимости коррекции гиперлипидемии у стероидчувсвительных пациентов, так как она купируется после наступления ремиссии.
Начатую в стационаре патогенетическую терапию необходимо продолжить в амбулаторных и домашних условиях пациента под ежемесячным контролем результатов лабораторных данных (ОАК, ОАМ, Биохимический анализ крови), кроме того:
− Мониторинг уровня протеинурии по тест-полоскам 1 раз в 1-2 недели, регулярное измерение АД.
− При нарастании протеинурии (рецидиве) определение протеин/креатининового коэффициента (для расчета суточной протеинурии) и коррекция иммуносупрессивной терапии;
− При резистентности к проводимой иммуносупрессивной терапии проведение повторной биопсии почки в условиях стационара.
Осложнения нефротического синдрома:
1. Инфекции. У пациентов с нефротическим синдромом могут быть разные инфекции: перитониты, целлюлиты, пневмония.
|
Инфекции |
Клиническая картина | Микроорганизмы | Терапия |
| Перитонит | Боли в животе, чувствительность при пальпации, рвота, диарея. В асцитической жикости более 100 лейкоцитов/мл, более 50% нейтрофилы | S. pneumoniae, S. pyogenes, E. coli | Цефотаксим или цефтриаксон 7-10 дней, ампициллин+аминогликозиды 7-10 дней |
| Пневмонии | Гипертермия, кашель, тахипное, крепитация | S. pneumoniae, H. influenzae, S. aureus | Внутрь: Амоксициллин, клавулановая кислота, эритромицин Парентерально: ампициллин+аминогликозиды или цефотаксим/цефтриаксон 7-10 дней |
| Целлюлит | Кожная эритема, уплотнение, болезненность | St aphylococci, Group A streptococci, H. influenzae | Амоксициллин, клавулановая кислота или цефтриаксон 7-10 дней |
| Грибковые инфекции | Легочная инфильтрация, длительная лихорадка, отсутствие ответа на антибактериальную терапию | Candida, Aspergillus spp. | Кожа, слизистые: Флуконазол 10 дней |
При наличии varicella zoster однократное введение иммуноглобулина 400мг/кг, внутривенно ацикловир (1500мг/м2/сут) 3 дня или внутрь 80мг/кг/сут 7-10 дней [16].
2. Тромбозы, имеется риск венозных, редко артериальных тромбозов. Применяются низкомолекулярные гепарины подкожно в течение длительного времени [17].
3. Гиповолемический шок бывает при назначении диуретиков, особенно когда имеется септицемия, диарея, рвота. Гиповолемический шок можно предположить при наличии разной интенсивности болей в животе, гипотензии, тахикардии, ознобе, в крови повышены уровни гематокрита, мочевины и мочевой кислоты. Восстанавливается путем срочных инфузий физиологических растворов из расчета 15-20мл/кг 20-30 минут, можно повторно. Инфузия 10% раствора альбумина (10мл/кг) или 20% раствора (0,5-1г/кг) при отсутствии эффекта после двух болюсов физ. раствора.
4. Побочные эффекты кортикостероидов: повышенный аппетит, задержка роста, риск инфекций, гипертензия, деминерализация костей, повышение глюкозы крови, катаракта
Медикаментозное лечение, оказываемое на амбулаторном уровне
Перечень основных лекарственных средств:
• метилпреднизолон, таблетки 4мг, 16мг;
• циклоспорин-А, капсулы 25мг, 50мг, 100мг;
• мофетила микофенолат, капсулы по 250мг;
• микофеноловая кислота, капсулы по 180мг;
• периндоприла аргинин, таблетки 5мг; 10мг;
• валсартан, таблетки 80мг, 160мг;
• торасемид, таблетки 5, 10, 20мг;
• индапамид, таблетки 1,25мг, 2,5мг
Перечень дополнительных лекарственных средств:
• кальция карбонат, магния карбонат, таблетки жевательные;
• фуросемид, таблетки 40мг;
• гидрохлортиазид, таблетки 25мг;
• спиронолактон, капсула 25мг, 50мг;
• флуконазол, капсулы 50мг;
• омепразол, капсулы 20мг;
• рабепразол, таблетки 20мг;
• ацикловир, таблетки 200мг, 400мг;
• амлодипин, таблетки 5мг, 10мг;
• карведилол, таблетки по 6,25мг, 12,5мг, 25мг;
• панангин, таблетки;
• эпоэтин-альфа, шприц-тюбики по 2000МЕ;
• эпоэтин-бета, шприц-тюбики по 2000МЕ.
Медикаментозное лечение, оказываемое на стационарном уровне
Перечень основных лекарственных средств:
• метилпреднизолон, таблетки 4мг,16мг, порошок для приготовления раствора для инъекций в комплекте с растворителем 250мг, 500 мг
• циклоспорин-А, капсулы 25мг, 50мг, 100мг
• мофетила микофенолат, капсулы по 250мг,
• микофеноловая кислота, капсулы по 180мг;
• циклофосфамид, порошок для приготовления раствора для внутривенного введения 200мг
• периндоприла аргинин, таблетки 5мг; 10мг;
• валсартан, таблетки 80мг, 160мг;
• торасемид, таблетки 5, 10, 20мг;
• индапамид, таблетки 1,25мг, 2,5мг
• лозаратан, таблетки 50мг
• валсартан, таблетки 80мг, 160мг
Перечень дополнительных лекарственных средств:
• ритуксимаб, флакон для внутривенных инфузий 100мг, 500мг
• иммуноглобулин человека нормальный, 10% раствор для инфузий 100мл
• месна, раствор для инъекций во флаконе 100 мг/мл 10,0мл
• ондансетрон, раствор для инъекций 4 мг/2мл, 8 мг/4мл
• кальция карбонат, магния карбонат, таблетки жевательные
• фуросемид, таблетки 40мг, раствор для инъекций 1% 2мл
• гидрохлортиазид, таблетки 25мг
• спиронолактон, капсула 25мг, 50мг
• флуконазол, капсулы 50мг
• омепразол, капсулы 20мг
• эпоэтин-альфа, шприц-тюбики по 2000МЕ
• эпоэтин-бета, шприц-тюбики по 2000МЕ
• ацикловир, таблетки 200мг, 400мг, порошок для приготовления раствора для инъекций 250мг
• надропарин, раствор для инъекций в предварительно наполненных шприцах, 2850 ме анти-ха/0,3мл
• цефотаксим, порошок для приготовления раствора для инъекций 1 г
• цефтриаксон, порошок для приготовления раствора для инъекций 1г,
• амоксициллин+клавулановая кислота, лиофилизат для приготовления раствора 625мг
• гепарин, раствор для инъекций 25000 ме/5мл
• альбумин, раствор для инфузий 10% 100мл, 20% 50мл
• амлодипин, таблетки 5мг, 10мг
• атенолол, таблетки по 50мг
• панангин, таблетки
• такролимус, таблетки по 0,5мг; 1мг;
• дексаметазон, таблетки по 500мкг;
• белимумаб, лиофилизат для приготовления раствора для инфузий, 120мг и 400мг
• инфликсимаб, порошок по 100мг для приготовления инфузий;
• адалимумаб, раствор для инфузий 40 мг/0,8 мл шприц, в комплекте с салфетками, № 1, № 2
• бортезомиб, порошок для приготовления раствора в/в 3,5 мг фл.
• колхицин, таблетки 1 мг
• гидрохлорохин, таблетки 200мг;
• альфакальцидол драже 0,25мкг; 0,5мкг.
Медикаментозное лечение, оказываемое на этапе скорой неотложной помощи:
При артериальной гипертензии (повышение АД более 160/100мм.рт.ст.) – фуросемид 1-2мг/кг внутривенно, нифедипин 5-10 мг под язык.
При почечной эклампсии – снижение АД, диазепам 1мл.
Другие виды лечения
Другие виды лечения, оказываемые на амбулаторном уровне: не проводятся.
Другие виды лечения, оказываемые на стационарном уровне:
Гемодиализ, изолированная ультрафильтрация показаны при почечной недостаточности острого периода с развитием симптомов:
• гиперкалиемия > 6,5ммоль/л;
• гипергидратация резистентная к диуретической терапии и риском развития отека легких;
• метаболический ацидоз, рН <7,2;
• анурия более 1 суток;
• уремия (мочевина > 30ммоль/л)
до выхода из критического состояния пациента.
Плазмаферез при ГУС, ТМА, системных васкулитах с повышением креатинина сыворотки крови > 500мкмоль/л, при криоглобулинемии, множественной миеломе, синдроме Гудпасчера – через день в течение 2-х недель непрерывным методом, с заменой 100% плазмы альбумином 10%.
Другие виды лечения, оказываемые на этапе скорой неотложной помощи: не проводятся.
Хирургическое вмешательство, оказываемое в стационарных условиях
В случаях возникновения необходимости проведения гемодиализа производится наложение стерильного одноразового двухпросветного катетера в центральные вены (внутренняя яремная, бедренная, подключичная).
Профилактические мероприятия:
• профилактика вирусных, бактериальных, грибковых инфекций – вакцинация против инфекций в неактивную стадию заболевания по показаниям;
• профилактика остеопороза – диета с большим содержанием кальция, лечебная физкультура, пероральный прием карбоната кальция, витамина Д;
• профилактика эклампсии, сердечно-сосудистой недостаточности – пероральный прием антигипертензивных препаратов, лечебная физкультура.
Дальнейшее ведение (сопровождение пациента на амбулаторном уровне):
Осмотр нефролога зависит от стадии заболевания:
— при активной стадии – 1 раз в месяц;
— при неактивной — 1 раз в 6 месяцев.
Контроль лабораторных данных в зависимости от стадии заболевания:
— в активной стадии заболевания: ОАК, ОАМ, креатинина, мочевины, АЛТ, АСТ, холестерина, глюкоза, определение белка в моче (количественная проба) 1 раз в месяц.
— в неактивной стадии заболевания: OAK, OAM, биохимический анализ крови (креатинин, мочевина, общий белок, холестерин, глюкоза), определение белка в моче (количественная проба) 1 раз в 6 месяцев, УЗИ почек 1 раз в год, ЭКГ-по показаниям
При ухудшении состояния пациента необходимо решение вопроса госпитализации.
Рекомендации по навыкам здорового образа жизни: коррекция факторов риска психопрофилактика, режим, диета.
Своевременная санация хронических очагов инфекции.
Ограничение физических нагрузок.
Исключение охлаждения и инсоляции.
Индикаторы эффективности лечения:
• купирование почечной недостаточности острого периода;
• достижение полной или частичной ремиссии заболевания (купирование/уменьшение отеков, купирование/уменьшение протеинурии до 0,5 г/сут, нормализация АД);
• отсутствие инфекционных и тромботических осложнений;
• замедление прогрессирования ХБП
Госпитализация
Показания для госпитализации
• дебют НС – для поиска возможных причин НС и верификации морфологического варианта поражения почек с последующим подбором этиотропной / патогенетической терапии;
• резистентность к стандартной иммуносупрессивной терапии.
Экстренная госпитализация:
• рецидив НС – для лечения рецидива и коррекции иммуносупрессивной терапии;
• осложнения – нефротический криз, инфекционные, тромботические, прогрессирующая почечная недостаточность.
Информация
Источники и литература
-
Протоколы заседаний Экспертного совета РЦРЗ МЗСР РК, 2014
- 1. Lameson J.L., Loscalzo J. Harrison’s Nephrology and Acid-Based Disorders. Edition 17th, 2010, New-York.
2. Клинические рекомендации. Стандарты ведения больных. Мухин Н.А., Козловская Л.В., Шилов Е.М. Выпуск 2, Гэотар Медиа, 2008 г, 1376 с.
3. Нефрология. Национальное руководство. Под ред. Мухина Н.А. Гэотар Медиа, 2008 г, 716 с.
4. Tune BM, Mendoza SA. Treatment of the idiopathic nephrotic syndrome: regimens and outcomes in children and adults. J Am Soc Nephrol 1997; 8:824.
5. Nolasco F, Cameron JS, Heywood EF, et al. Adult-onset minimal change nephrotic syndrome: a long-term follow-up. Kidney Int 1986; 29:1215.
6. Fujimoto S, Yamamoto Y, Hisanaga S, et al. Minimal change nephrotic syndrome in adults: response to corticosteroid therapy and frequency of relapse. Am J Kidney Dis 1991; 17:687.
7. Nakayama M, Katafuchi R, Yanase T, et al. Steroid responsiveness and frequency of relapse in adult-onset minimal change nephrotic syndrome. Am J Kidney Dis 2002; 39:503.
8. Korbet SM, Schwartz MM, Lewis EJ. Minimal-change glomerulopathy of adulthood. Am J Nephrol 1988; 8:291.
9. Mak SK, Short CD, Mallick NP. Long-term outcome of adult-onset minimal-change nephropathy. Nephrol Dial Transplant 1996; 11:2192.
10.Arneil GC, Lam CN. Long-term assessment of steroid therapy in childhood nephrosis. Lancet 1966; 2:819.
11.Tse KC, Lam MF, Yip PS, et al. Idiopathic minimal change nephrotic syndrome in older adults: steroid responsiveness and pattern of relapses. Nephrol Dial Transplant 2003; 18:1316.
12.Meyrier A. Nephrotic focal segmental glomerulosclerosis in 2004: an update. Nephrol Dial Transplant 2004; 19:2437.
13.Gipson DS, Trachtman H, Kaskel FJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int 2011; 80:868.
14.Muso E, Mune M, Yorioka N, et al. Beneficial effect of low-density lipoprotein apheresis (LDL-A) on refractory nephrotic syndrome (NS) due to focal glomerulosclerosis (FGS). Clin Nephrol 2007; 67:341.
15.Joy MS, Gipson DS, Dike M, et al. Phase I trial of rosiglitazone in FSGS: I. Report of the FONT Study Group. Clin J Am Soc Nephrol 2009; 4:39.
16.Hausmann R, Kuppe C, Egger H, et al. Electrical forces determine glomerular permeability. J Am Soc Nephrol 2010; 21:2053.
17.Reiser J, von Gersdorff G, Loos M, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest 2004; 113:1390.
18.Schönenberger E, Ehrich JH, Haller H, Schiffer M. The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant 2011; 26:18.
19.Gbadegesin R, Lavin P, Foreman J, Winn M. Pathogenesis and therapy of focal segmental glomerulosclerosis: an update. Pediatr Nephrol 2011; 26:1001.
20.Chen T, Lv Y, Lin F, Zhu J. Acute kidney injury in adult idiopathic nephrotic syndrome. Ren Fail 2011; 33:144.
21.Frisch G, Lin J, Rosenstock J, et al. Mycophenolate mofetil (MMF) vs placebo in patients with moderately advanced IgA nephropathy: a double-blind randomized controlled trial. Nephrol Dial Transplant 2005; 20:2139.
22.Le W, Liang S, Hu Y, et al. Long-term renal survival and related risk factors in patients with IgA nephropathy: results from a cohort of 1155 cases in a Chinese adult population. Nephrol Dial Transplant 2012; 27:1479.
23.Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30.
24.Orlowski T, Rancewicz Z, Lao M, et al. Long-term immunosuppressive therapy of idiopathic membranoproliferative glomerulonephritis. Klin Wochenschr 1988; 66:1019.
25.Cattran DC, Cardella CJ, Roscoe JM, et al. Results of a controlled drug trial in membranoproliferative glomerulonephritis. Kidney Int 1985; 27:436.
26.Faedda R, Satta A, Tanda F, et al. Immunosuppressive treatment of membranoproliferative glomerulonephritis. Nephron 1994; 67:59.
27.Bhat P, Weiss S, Appel GB, Radhakrishnan J. Rituximab treatment of dysproteinemias affecting the kidney: a review of three cases. Am J Kidney Dis 2007; 50:641.
28.Vilayur E, Trevillian P, Walsh M. Monoclonal gammopathy and glomerulopathy associated with chronic lymphocytic leukemia. Nat Clin Pract Nephrol 2009; 5:54.
29.Dillon JJ, Hladunewich M, Haley WE, et al. Rituximab therapy for type I membranoproliferative glomerulonephritis. Clin Nephrol 2012; 77:290.
30.Jones G, Juszczak M, Kingdon E, et al. Treatment of idiopathic membranoproliferative glomerulonephritis with mycophenolate mofetil and steroids. Nephrol Dial Transplant 2004; 19:3160.
31.Choi MJ, Eustace JA, Gimenez LF, et al. Mycophenolate mofetil treatment for primary glomerular diseases. Kidney Int 2002; 61:1098.
32.Bayazit AK, Noyan A, Cengiz N, Anarat A. Mycophenolate mofetil in children with multidrug-resistant nephrotic syndrome. Clin Nephrol 2004; 61:25.
33.Bagheri N, Nemati E, Rahbar K, et al. Cyclosporine in the treatment of membranoproliferative glomerulonephritis. Arch Iran Med 2008; 11:26.
34.Cattran DC. Current status of cyclosporin A in the treatment of membranous, IgA and membranoproliferative glomerulonephritis. Clin Nephrol 1991; 35 Suppl 1:S43.
35.Matsumoto H, Shibasaki T, Ohno I, et al. [Effect of cyclosporin monotherapy on proteinuria in a patient with membranoproliferative glomerulonephritis]. Nihon Jinzo Gakkai Shi 1995; 37:258.
36.Haddad M, Lau K, Butani L. Remission of membranoproliferative glomerulonephritis type I with the use of tacrolimus. Pediatr Nephrol 2007; 22:1787.
37.Donadio JV Jr, Anderson CF, Mitchell JC 3rd, et al. Membranoproliferative glomerulonephritis. A prospective clinical trial of platelet-inhibitor therapy. N Engl J Med 1984; 310:1421.
38.Zäuner I, Böhler J, Braun N, et al. Effect of aspirin and dipyridamole on proteinuria in idiopathic membranoproliferative glomerulonephritis: a multicentre prospective clinical trial. Collaborative Glomerulonephritis Therapy Study Group (CGTS). Nephrol Dial Transplant 1994; 9:619.
39.Zimmerman SW, Moorthy AV, Dreher WH, et al. Prospective trial of warfarin and dipyridamole in patients with membranoproliferative glomerulonephritis. Am J Med 1983; 75:920.
40.Harmankaya O, Baştürk T, Oztürk Y, et al. Effect of acetylsalicylic acid and dipyridamole in primary membranoproliferative glomerulonephritis type I. Int Urol Nephrol 2001; 33:583.
41.Servais A, Frémeaux-Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 2007; 44:193.
42.Habbig S, Mihatsch MJ, Heinen S, et al. C3 deposition glomerulopathy due to a functional factor H defect. Kidney Int 2009; 75:1230.
43.Fakhouri F, Frémeaux-Bacchi V, Noël LH, et al. C3 glomerulopathy: a new classification. Nat Rev Nephrol 2010; 6:494.
44.Radhakrishnan S, Lunn A, Kirschfink M, et al. Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med 2012; 366:1165.
45.Bomback AS, Smith RJ, Barile GR, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 2012; 7:748.
46.Licht C, Weyersberg A, Heinen S, et al. Successful plasma therapy for atypical hemolytic uremic syndrome caused by factor H deficiency owing to a novel mutation in the complement cofactor protein domain 15. Am J Kidney Dis 2005; 45:415.
47. Клинические практические рекомендации KDIGO по лечению гломерулонефритов, 2012г. Нефрология и диализ, 2014г. приложение.
- 1. Lameson J.L., Loscalzo J. Harrison’s Nephrology and Acid-Based Disorders. Edition 17th, 2010, New-York.
Информация
III. ОРГАНИЗАЦИОННЫЕ АСПЕКТЫ ВНЕДРЕНИЯ ПРОТОКОЛА
Список разработчиков:
1) Туганбекова Салтанат Кенесовна – доктор медицинских наук, профессор АО «Национальный научный медицинский центр», заместитель генерального директора по науке, главный внештатный нефролог Министерства здравоохранения и социального развития Республики Казахстан
2) Гайпов Абдужаппар Эркинович – кандидат медицинских наук АО «Национальный научный медицинский центр» руководитель отдела экстракорпоральной гемокоррекции, врач-нефролог
3) Туребеков Думан Кажибаевич – доктор медицинских наук, ГКП на ПХВ «Городская больница №1» Управления здравоохранения города Астаны, заведующий отделением нефрологии, главный внештатный нефролог города Астаны
4) Султанова Багдат Газизовна – доктор медицинских наук, профессор АО «Казахский медицинский университет непрерывного образования», заведующая кафедрой нефрологии и гемодиализа
5) Жусупова Гульнар Даригеровна – кандидат медицинских наук АО «Медицинский университет Астана», врач-клинический фармаколог, ассистент кафедры общей и клинической фармакологии
Указание на отсутствие конфликта интересов: отсутствует.
Рецензенты:
Кабулбаев Кайрат Абдуллаулы – доктор медицинских наук, профессор РГП на ПХВ «Казахский национальный медицинский университет имени С.Д. Асфендиярова», руководитель модуля нефрологии
Указание условий пересмотра протокола: пересмотр протокола через 3 года и/или при появлении новых методов диагностики/ лечения с более высоким уровнем доказательности.
Прикреплённые файлы
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники. Стандарты лечения
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Внимание!
Если вы не являетесь медицинским специалистом:
-
Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
-
Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
-
Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
-
Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
-
Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.