Синдром Ди Джорджи

Синдром Ди Джорджи – генетическое заболевание, относящееся к группе первичных иммунодефицитов и, наряду с ослаблением иммунитета, характеризующееся многочисленными пороками развития. Симптомами этого состояния являются частые бактериальные инфекции со склонностью к тяжелому течению, врожденные пороки сердца, аномалии развития лица и другие нарушения. Диагностика синдрома Ди Джорджи основывается на исследовании сердца, щитовидных и паращитовидных желез, изучении иммунологического статуса и данных молекулярно-генетических анализов. Лечение только симптоматическое, включает хирургическую коррекцию пороков сердца и аномалий лица, заместительную иммунологическую терапию, борьбу с бактериальными и грибковыми инфекциями.
Общие сведения
Синдром Ди Джорджи (гипоплазия тимуса и паращитовидных желез, велокардиофациальный синдром) – генетическое заболевание, обусловленное нарушением эмбрионального развития третьего и четвертого фарингеальных мешков. Впервые это состояние было описано в 1965 году американским педиатром Анджело Ди Джорджи, который классифицировал его как врожденную аплазию тимуса и паращитовидных желез. Дальнейшие исследования в области генетики помогли определить, что нарушения при этом заболевании выходят далеко за рамки первичного иммунодефицита. Это дало основание для появления другого названия синдрома Ди-Джорджи. С учетом наиболее часто поражаемых органов (небо, сердце, лицо) некоторые специалисты именуют данную патологию велокардиофациальным синдромом. Ряд современных исследователей разграничивают эти два состояния и считают, что «истинный» велокардиофациальный синдром не сопровождается выраженными иммунологическими нарушениями. Встречаемость синдрома Ди Джорджи составляет 1:3 000-20 000 – такое значительное расхождение данных обусловлено тем, что достоверная и четкая граница между этим заболеванием и велокардиофациальным синдромом до сих пор не установлена. Поэтому один и тот же больной, по мнению разных специалистов, может иметь либо первичный иммунодефицит, сопровождающийся сопутствующими нарушениями, либо многочисленнее пороки развития на фоне снижения иммунитета.

Синдром Ди Джорджи
Причины синдрома Ди Джорджи
Генетическая природа синдрома Ди Джорджи заключается в повреждении центральной части длинного плеча 22-й хромосомы, где предположительно располагаются гены, кодирующие ряд важных факторов транскрипции. Удалось идентифицировать один из этих генов – TBX1, продуктом его экспрессии является белок под названием T-box. Он относится к семейству протеинов, контролирующих процессы эмбриогенеза. Доказательством взаимосвязи синдрома Ди Джорджи и TBX1 является тот факт, что незначительный процент больных не имеет выраженных повреждений 22-й хромосомы, присутствуют только мутации в этом гене. Также высказываются предположения о роли делеций иных хромосом в развитии данного заболевания. Так, аналогичные синдрому Ди Джорджи проявления выявлялись при наличии повреждений 10-й, 17-й и 18-й хромосом.
В большинстве случаев синдрома Ди Джорджи делеция 22-й хромосомы захватывает порядка 2-3 миллионов пар оснований. Чаще всего данный генетический дефект возникает спонтанно во время формирования мужских или женских половых клеток – то есть, носит герминативный характер. Лишь десятая часть всех случаев заболевания представляет собой семейную форму с аутосомно-доминантным характером наследования. Патогенез синдрома Ди Джорджи сводится к нарушению формирования особых эмбриональных образований – фарингеальных мешков (главным образом, 3-го и 4-го), которые являются предшественниками ряда тканей и органов. Главным образом, они отвечают за формирование неба, паращитовидных желез, тимуса, сосудов средостения и сердца, поэтому при синдроме Ди Джорджи возникают пороки развития именно этих органов.
Симптомы синдрома Ди Джорджи
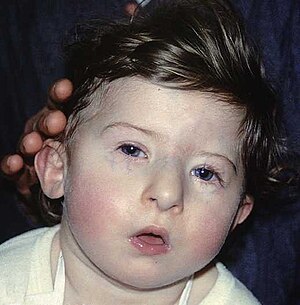
Многие проявления синдрома Ди Джорджи определяются сразу после рождения ребенка, отдельные пороки развития (например, сердца) можно выявить еще раньше – на профилактических ультразвуковых исследованиях. Чаще всего первыми обнаруживаются аномалии развития лица – расщепление неба, иногда в сочетании с «заячьей губой», прогнатия нижней челюсти. Зачастую младенцы с синдромом Ди Джорджи имеют небольшой рот, маленький нос с расширенной переносицей, деформированные или недоразвитые хрящи ушных раковин. При относительно легком течении заболевания все вышеперечисленные симптомы могут быть выражены довольно слабо, даже расщепление твердого неба может возникать только в задней его части и выявляться лишь при тщательном осмотре у отоларинголога.
В первые месяцы жизни больного синдромом Ди Джорджи на первый план выступают проявления врожденных пороков сердца – это может быть как тетрада Фалло, так и отдельные нарушения: дефект межжелудочковой перегородки, незаращение артериального протока и ряд других. Они сопровождаются цианозом, сердечно-сосудистой недостаточностью и при отсутствии квалифицированной медицинской помощи (в том числе и хирургической) могут приводить к ранней смерти больных. Другим распространенным нарушением у детей с синдромом Ди Джорджи считаются судороги и тетания, обусловленная гипоплазией паращитовидных желез и последующей гипокальциемией.
Следующим важнейшим проявлением синдрома Ди Джорджи, отличающим его от других разновидностей велокардиофациального синдрома, является выраженный первичный иммунодефицит. Он развивается по причине аплазии или недоразвития тимуса и поэтому в большей степени затрагивает клеточный иммунитет. Однако из-за тесных взаимосвязей между гуморальным и клеточным отделами иммунной системы это приводит к общему ослаблению защитных сил организма. Больные с синдромом Ди Джорджи крайне чувствительны к вирусным, грибковым и бактериальным инфекциям, которые нередко принимают затяжное и тяжелое течение. Некоторые исследователи отмечают наличие умственной отсталости различной степени, иногда могут наблюдаться судороги неврологического происхождения.
Диагностика синдрома Ди Джорджи
Для определения синдрома Ди Джорджи применяют метод физикального общего осмотра, кардиологические исследования (ЭхоКГ, электрокардиограмма), УЗИ щитовидной железы и тимуса, иммунологические пробы. Вспомогательную роль играет проведение общего и биохимического анализов крови, изучение анамнеза больного, генетические исследования. При осмотре больных синдромом Ди Джорджи могут определяться характерные для заболевания нарушения – расщепление твердого неба, аномалии строения лица, патологии ЛОР-органов. В анамнезе, как правило, выявляются частые эпизоды вирусных и грибковых инфекций, принимающих тяжелое течение, судороги, обусловленные гипокальциемией, нередко обнаруживается обширное кариозное поражение зубов.
На ультразвуковых исследованиях вилочковой железы отмечается значительное уменьшение массы или даже полное отсутствие органа (агенезия). ЭхоКГ и другие кардиологические методы диагностики выявляют многочисленные пороки сердца (например, дефект межжелудочковой перегородки) и сосудов средостения. Иммунологические исследования подтверждают значительное падение уровня Т-лимфоцитов. Это же явление наблюдается в периферической крови и нередко сочетается с уменьшением концентрации белков-иммуноглобулинов. Биохимическое изучение крови свидетельствует о снижении уровня кальция и гормонов паращитовидной железы. Врач-генетик может выполнить поиск делеций в 22-й хромосоме посредством флуоресцентной гибридизации ДНК или мультиплексной полимеразной цепной реакции.
Лечение синдрома Ди Джорджи
Специфического лечения синдрома Ди Джорджи на сегодняшний момент не существует, используют только паллиативные и симптоматические методики. Очень важно как можно раньше выявить врожденные пороки сердца и при необходимости произвести их хирургическую коррекцию, поскольку именно сердечно-сосудистые нарушения являются наиболее частой причиной неонатальной смерти при этом заболевании. Значительную опасность представляют собой судорожные приступы, обусловленные гипокальциемией, что требует своевременной коррекции электролитного баланса плазмы крови. Помощь хирургов при синдроме Ди Джорджи также может потребоваться для устранения пороков развития лица и неба.
Из-за выраженного иммунодефицита любые признаки бактериальной, вирусной или грибковой инфекции являются поводом для срочного применения соответствующих препаратов (антибиотиков, противовирусных и фунгицидных средств). Для улучшения иммунного статуса больного синдромом Ди Джорджи может производиться заместительное вливание иммуноглобулинов, полученных из донорской плазмы. В отдельных случаях осуществлялась пересадка вилочковой железы, которая стимулировала образование собственных Т-лимфоцитов – это способствовало улучшению качества жизни больных.
Прогноз и профилактика синдрома Ди Джорджи
Прогноз синдрома Ди Джорджи большинством исследователей оценивается как неопределенный, так как данное заболевание характеризуется значительной вариабельностью симптомов. В тяжелых случаях имеется высокий риск ранней неонатальной смерти из-за сочетания сердечно-сосудистых и иммунологических нарушений. Более доброкачественные формы синдрома Ди Джорджи требуют достаточно интенсивной паллиативной терапии, особенно важно уделять внимание лечению и профилактике вирусных и грибковых инфекций. Интеллектуальное развитие больных несколько замедлено, однако при правильной педагогической и психологической коррекции проявления задержки развития можно нивелировать. Из-за частого спонтанного характера мутаций профилактика синдрома Ди Джорджи не разработана.
Синдром Ди Джорджи — лечение в Москве
Общие сведения
Синдром Ди Джорджи еще в некоторых источниках указывают как синдром Ди Георга. Впервые патология была описана в 1965 году американским педиатром и эндокринологом — Анджело Мария Ди Джорджи (фото). По международной классификации десятого пересмотра (МКБ-10) данному иммунодефициту был присвоен код D82.1 и указан как синдром дивертикула глотки, вилочковой железы: алимфоплазия, аплазия либо гипоплазия с иммунной недостаточностью.
")
Angelo DiGeorge(15.04.1921-11.10.2009)
Синдром Ди Джорджа как первичный иммунодефицит является редким врожденным заболеванием (1 случай на 4-6 тыс. человек) и относится к идиопатическому изолированному гипопаратиреозу – состоянию, для которого характерно снижение выработки паратгормонов и гипокальциемия. Для синдрома свойственно множество фенотипов поражения, осложнений и присоединения сопутствующих нарушений.
Патогенез
Для синдрома Ди Георга характерна аплазия или гипоплазия вилочковой железы (тимуса), а также агенезия или дисгенезия паращитовидных желез в результате нарушений закладки и эмбриональной дифференцировки 3-4 жаберных (глоточных) карманов. Это вызывает резкое снижение популяции Т-лимфоцитов, нарушение их дифференцировки и как следствие — иммунологическую недостаточность. У детей могут развиваться различные врождённые аномалии крупных сосудов, например, дефекты аорты, межжелудочковой перегородки или синий порок сердца.

Расположение паращитовидных и щитовидных желез
Классификация
В зависимости от спектра клинических проявлений и врожденных аномалий синдром Ди Георга может быть полным и частичным, к примеру, протекать как изолированная недостаточность паращитовидных желёз либо врождённое отсутствие паращитовидных (околощитовидных) желез, которые приводят к гипокальциемическим судорогам, наблюдающимся у новорожденных в виде неонатальной тетании. Нарушения закладки тимуса приводят к развитию различных инфекционных заболеваний и обычно вызваны иммунологической недостаточностью.
Достаточно часто болезнь протекает в менее тяжелой форме и является наиболее распространенной причиной умственной отсталости с малым количеством других проявлений, поэтому может быть формально не диагностирована.
Причины
У синдрома Ди Джорджи или врождённой аплазии тимуса и паращитовидной железы генетическая причина развития. Патология развивается в результате делеции центрального участка более длинного плеча двадцать второй хромосомы, поэтому еще обозначается как синдром 22q 11.2. Однако, в некоторых случаях аналогичная клиническая картина наблюдалась и после других хромосомных перестроек – транслокаций и микроделеций, развивающихся в таких хромосомах как ТВХ1, 10р13, 17р13, 18q21 и пр. Эти мутации спорадические в 90% и обычно происходят на этапе мейоза спермато— или овогенеза.
Для данного синдрома дисэмбриогенеза 3-4 жаберной дуги — фенотипа CATCH 22 характерно наследование по аутосомно-доминантному типу, которое наблюдается лишь в 5-10% случаев, но при этом не исключается возможность аутосомно-рецессивного типа наследования с разной экспрессивностью.
Симптомы врождённой аплазии тимуса и паращитовидных желёз (Синдром Ди Джорджи)
Синдром Ди Джорджи – это первичный иммунодефицит, вызванный врожденным отсутствием или наличием аномалий развития тимуса и паращитовидных желез, который отличается триадой клинических симптомов, включающей:
- различные врожденные пороки сердца, дефекты крупных сосудов, носа, рта, ушей, что в результате дает специфические черты лица (как на фото): гипертелоризм (увеличенное расстояние между глазами), микрогнатия (недоразвитие нижней челюсти), низкое расположение ушных раковин;
- первичный иммунодефицит – нарушен как клеточный, так и гуморальный ответ иммунитета в результате аплазии вилочковой железы и невозможности обеспечения нормального развития Т-клеток;
- прогрессирующая гипокальциемия (пониженная концентрация кальция в кровотоке) – вызвана гипопаратиреозом, сопровождается судорожным синдромом и развитием скелетных аномалий.

Ребенок со специфическими чертами, характерными для синдрома Ди Джорджи
Особенности синдрома могут широко варьироваться даже в пределах одной семьи и затрагивать различные системы органов. Сопутствующими проблемами становится:
- нарушение работы почек и их атрофия, развитие нефрокальциноза, гидронефроза и пр.;
- проблемы с пищеварением и перистальтикой ЖКТ;
- дефицит гормона роста;
- проблемы с речью;
- психические расстройства, в том числе шизофрения;
- потеря слуха (проводящая и нейросенсорная обычно вызвана черепно-лицевыми синдромами);
- нарушения координации, которые могут быть вызваны гипоплазией мозжечка;
- синюшность кожных покровов в результате плохого кровообращения;
- ревматоидный артрит;
- частые переломы костей;
- болезнь Грейвса;
- болезнь Паркинсона.
Анализы и диагностика
Для подтверждения диагноза синдром Ди Джорджа необходимо выявление агенезии или дисгенеза паращитовидных желёз, аплазии вилочковой железы, иммунологической недостаточности, черепно-лицевых дисморфий (микрогнатии, гипертелоризма, антимонголоидного разреза глаз, расщелины губы и нёба, деформированных и/или низко расположенных ушных раковин) и прочих типичных аномалий развития. При этом оказываются эффективными различные исследования, включая УЗИ, МРТ и пр., а также используются методы генетического тестирования, к примеру, FISH (метод флуоресцентной гибридизации in situ).
Наиболее яркими проявлениями является гипопаратериоз и молочница. Также при плановом обследовании выявляются дефекты аорты, тетрада Фалло, катаракта, паховая грыжа и тому подобное. При помощи серологических исследований можно выявить лимфоцитопению, гипокальциемию, гипоγ-глобулинемии. Благодаря иммунологическим методам удается обнаружить нарушения трансформации лимфоцитов и их функциональной активности, в 20% случаев наблюдается сниженное количество Т-клеток. После иммунологических исследований важно провести дифференциальный диагноз с прочими первичными иммунодефицитами, таким как: синдром Вискотта-Олдрича, Брутона.
Лечение
Так как недуг неизлечим больным обычно назначают симптоматическое и комплексное поддерживающее лечение. Основными приемами считается соблюдение диеты, стерильные условия, прием кальция, комплексов витамин, антибиотиков, противогрибковых, иммуномодулирующих, седативных препаратов и пр.
Доктора
Лекарства
В зависимости от особенностей течения и имеющихся проблем со здоровьем могут быть назначены:
- противовирусные, противогрибковые препараты, а также антибиотики широкого спектра действия для лечения инфекционных заболеваний;
- пожизненный прием витамина Д и кальция;
- заместительная терапия иммуноглобулином.
Процедуры и операции
Для наилучшего прогноза пациентов с синдромом Ди Георга необходимо раннее выявление всех аномалий развития и их устранение стандартными методами лечения:
- трансплантация эмбриональных тканей тимуса;
- кардиохирургия при врожденных пороках сердца.
Первичный иммунодефицит у детей
В результате клеточного иммунодефицита дети с синдромом Ди Джорджи очень подвержены действию инфекции вирусной, грибковой и некоторых разновидностей бактериальной природы. Чаще всего развивается затяжной и тяжелый ринит, пневмония, абсцесс, пиелонефрит, колит, сепсис и т.д. Если ребенку удается пережить пятилетний возраст, то может не обнаруживаться недостаточность Т-клеток, так как антиген-независимый этап созревания Т-клеток начинает происходить не в тимусе, а в многослойном плоском эпителии.
У новорожденных часто возникают судороги и проблемы с кормлением. Младенцы слабые, у них плохой аппетит. Нарушение перистальтики приводит к частым запором. В дальнейшем у деток наблюдается задержка роста и развития, возникают проблемы с обучением, а также когнитивные нарушения, например, приобретение аутистических черт поведения.
У детей с синдромом ДиДжорджа специфический профиль, выявляемый нейропсихологическими тестами. У них нормальный IQ и они способны проходить обучение в обычной школе, дома или заниматься в специальных классах, ведь очень часто у больных есть проблемы с речью (неразборчивость, ошибки артикуляции, сложности при приобретении словарного запаса).
Прогноз синдрома Ди Джорджи
К сожалению, не сегодняшний день, современная медицина оказывается бессильна перед данным генетическим заболеванием. Прогноз крайне неблагоприятный – дети умирают от различных инфекционных заболеваний или сердечной недостаточности, поэтому лучшим способом предупреждения развития врожденных патологий считается медико-генетическое консультирование и пренатальная диагностика наследственных болезней.
Список источников
- Стефани Д.В., Вельтищев Ю.Е. Иммунология и иммунопатология детского возраста.— М., 1996, -С. 27.
- Ивановская Т.Е., Цинзерлинг А.В. Патологическая анатоми.— М., 1976. -С. 136.
From Wikipedia, the free encyclopedia
| DiGeorge syndrome | |
|---|---|
| Other names | DiGeorge anomaly,[1][2] velocardiofacial syndrome (VCFS),[3] Shprintzen syndrome,[4] conotruncal anomaly face syndrome (CTAF),[5] Takao syndrome,[6] Sedlackova syndrome,[7] Cayler cardiofacial syndrome,[7] CATCH22,[7] 22q11.2 deletion syndrome[7] |
 |
|
| A child with characteristic facial features of DiGeorge syndrome | |
| Specialty | Medical genetics |
| Symptoms | Varied; commonly congenital heart problems, specific facial features, cleft palate[7] |
| Complications | Kidney problems, hearing loss, autoimmune disorders[7] |
| Causes | Genetic (typically new mutation)[7] |
| Diagnostic method | Based on symptoms and genetic testing[5] |
| Differential diagnosis | Smith–Lemli–Opitz syndrome, Alagille syndrome, VACTERL, Oculo-auriculo-vertebral spectrum[5] |
| Treatment | Involves many healthcare specialties[5] |
| Prognosis | Depends on the specific symptoms[3] |
| Frequency | 1 in 4,000[7] |
DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22.[7] While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental delay, intellectual disability and cleft palate.[7] Associated conditions include kidney problems, schizophrenia, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves’ disease.[7][8]
DiGeorge syndrome is typically due to the deletion of 30 to 40 genes in the middle of chromosome 22 at a location known as 22q11.2.[3] About 90% of cases occur due to a new mutation during early development, while 10% are inherited.[7] It is autosomal dominant, meaning that only one affected chromosome is needed for the condition to occur.[7] Diagnosis is suspected based on the symptoms and confirmed by genetic testing.[5]
Although there is no cure, treatment can improve symptoms.[3] This often includes a multidisciplinary approach with efforts to improve the function of the potentially many organ systems involved.[9] Long-term outcomes depend on the symptoms present and the severity of the heart and immune system problems.[3] With treatment, life expectancy may be normal.[10]
DiGeorge syndrome occurs in about 1 in 4,000 people.[7] The syndrome was first described in 1968 by American physician Angelo DiGeorge.[11][12] In late 1981, the underlying genetics were determined.[12]
Signs and symptoms[edit]
The features of this syndrome vary widely, even among members of the same family, and affect many parts of the body. Characteristic signs and symptoms may include birth defects such as congenital heart disease, defects in the palate, most commonly related to neuromuscular problems with closure (velopharyngeal insufficiency), learning disabilities, mild differences in facial features, and recurrent infections. Infections are common in children due to problems with the immune system’s T cell-mediated response that in some patients is due to an absent or hypoplastic thymus. DiGeorge syndrome may be first spotted when an affected newborn has heart defects or convulsions from hypocalcemia due to malfunctioning parathyroid glands and low levels of parathyroid hormone (parathormone).[citation needed]
Affected individuals may also have other kinds of birth defects including kidney abnormalities and significant feeding difficulties as babies. Gastrointestinal issues are also very common in this patient population. Digestive motility issues may result in constipation.[13] Disorders such as hypothyroidism and hypoparathyroidism or thrombocytopenia (low platelet levels), and psychiatric illnesses are common late-occurring features.[14]
Microdeletions in chromosomal region 22q11.2 are associated with a 20 to 30-fold increased risk of schizophrenia.[15] Studies provide various rates of 22q11.2DS in schizophrenia, ranging from 0.5 to 2.0% and averaging about 1.0%, compared with the overall estimated 0.025% risk of the 22q11.2DS in the general population.[16]
Salient features can be summarized using the mnemonic CATCH-22 to describe 22q11.2DS, with the 22 signifying the chromosomal abnormality is found on the 22nd chromosome, as below:[17]
- Cardiac abnormality (commonly interrupted aortic arch, truncus arteriosus and tetralogy of Fallot)
- Abnormal facies
- Thymic aplasia or hypoplasia
- Cleft palate
- Hypocalcemia/hypoparathyroidism early in life
Individuals can have many possible features, ranging in number of associated features and from the mild to the very serious. Symptoms shown to be common include:
- Congenital heart disease (40% of individuals), particularly conotruncal malformations (interrupted aortic arch (50%), persistent truncus arteriosus (34%), tetralogy of Fallot, and ventricular septal defect)
- Cyanosis (bluish skin due to poor circulation of oxygen-rich blood)
- Palatal abnormalities (50%), particularly velopharyngeal incompetence, submucosal cleft palate, and cleft palate; characteristic facial features (present in the majority of Caucasian individuals) including hypertelorism
- Learning difficulties (90%), including cognitive deficits, attention deficit disorders[18]
- Hypocalcemia (50%)(due to hypoparathyroidism)
- Significant feeding problems (30%)
- Renal anomalies (37%)
- Hearing loss (both conductive and sensorineural) (hearing loss with craniofacial syndromes)
- Laryngotracheoesophageal anomalies
- Growth hormone deficiency
- Autoimmune disorders
- Immune disorders due to reduced T cell numbers
- Schizophrenia develops in 25-30% by adulthood
- Seizures (with or without hypocalcemia)
- Skeletal abnormalities
- Psychiatric disorders[18]
This syndrome is characterized by incomplete penetrance. Therefore, there is a marked variability in clinical expression between the different patients. This often makes early diagnosis difficult.[19]
Cognitive impairments[edit]
Children with DiGeorge syndrome have a specific profile in neuropsychological tests. They usually have a below-borderline normal IQ, with most individuals having higher scores in the verbal than the nonverbal domains. Some are able to attend mainstream schools, while others are home-schooled or in special classes. The severity of hypocalcemia early in childhood is associated with autism-like behavioral difficulties.[20]
Adults with DiGeorge syndrome are a specifically high-risk group for developing schizophrenia. About 30% have at least one episode of psychosis and about a quarter develop schizophrenia by adulthood.[21]
Individuals with DiGeorge syndrome also have a higher risk of developing early onset Parkinson’s disease (PD). Diagnosis of Parkinson’s can be delayed by up to 10 years due to the use of antipsychotics, which can cause parkinsonian symptoms.[22][23]
Speech and language[edit]
Current research demonstrates a unique profile of speech and language impairments is associated with 22q11.2DS. Children often perform lower on speech and language evaluations in comparison to their nonverbal IQ scores.[contradictory] Common problems include hypernasality, language delays, and speech sound errors.[24][25][26]
Hypernasality occurs when air escapes through the nose during the production of oral speech sounds, resulting in reduced intelligibility. This is a common characteristic in the speech and language profile because 69% of children have palatal abnormalities. If the structure of the soft palate velum is such that it does not stop the flow of air from going up to the nasal cavity, it will cause hypernasal speech. This phenomenon is referred as velopharyngeal inadequacy (VPI). Hearing loss can also contribute to increased hypernasality because children with hearing impairments can have difficulty self monitoring their oral speech output. The treatment options available for VPI include prosthesis and surgery.[24][25][27][28][29]
Difficulties acquiring vocabulary and formulating spoken language (expressive language deficits) at the onset of language development are also part of the speech and language profile associated with the 22q11.2 deletion. Vocabulary acquisition is often severely delayed for preschool-age children. In some recent studies, children had a severely limited vocabulary or were still not verbal at 2–3 years of age. School-age children do make progress with expressive language as they mature, but many continue to have delays and demonstrate difficulty when presented with language tasks such as verbally recalling narratives and producing longer and more complex sentences. Receptive language, which is the ability to comprehend, retain, or process spoken language, can also be impaired, although not usually with the same severity as expressive language impairments.[25][28][29]
Articulation errors are commonly present in children with DiGeorge syndrome. These errors include a limited phonemic (speech sound) inventory and the use of compensatory articulation strategies resulting in reduced intelligibility. The phonemic inventory typically produced consists of sounds made in the front or back of the oral cavity such as: /p/, /w/, /m/, /n/, and glottal stops. Sound made in the middle of the mouth are completely absent. Compensatory articulation errors made by this population of children include: glottal stops, nasal substitutions, pharyngeal fricatives, linguapalatal sibilants, reduced pressure on consonant sounds, or a combination of these symptoms. Of these errors, glottal stops have the highest frequency of occurrence. It is reasoned that a limited phonemic inventory and the use of compensatory articulation strategies is present due to the structural abnormalities of the palate. The speech impairments exhibited by this population are more severe during the younger ages and show a trend of gradual improvement as the child matures.[24][28]
Genetics[edit]

DiGeorge syndrome is caused by a heterozygous deletion of part of the long arm (q) of chromosome 22, region 1, band 1, sub-band 2 (22q11.2). Approximately 80-90% of patients have a deletion of 3 Mb and 8% have a deletion of 1.5Mb.[31][32] The number of genes affected by the deletion has been cited as approximately 30 to 50.[33][34] Very rarely, patients with somewhat similar clinical features may have deletions on the short arm of chromosome 10.[35] The disorder has an autosomal dominant inheritance pattern.
A French study of 749 people diagnosed between 1995 and 2013 found that the mutation was inherited in 15% of patients, of which 85.5% was from the mother.[36] Other studies have found inheritance rates of 6-10%. The majority cases are a result of a de novo (new to the family) deletion.[13] This is because the 22q11 region has a structure that makes it highly prone to rearrangements during sperm or egg formation.[37]
The exact mechanism that causes all of the associated features of the syndrome is unknown.[31] Of the 30–50 genes in the deleted region, a number have been identified as possibly playing a role in the development of some of the signs and symptoms.
TBX1[edit]
Haploinsufficiency of the TBX1 gene (T-box transcription factor TBX1) is thought to be the cause of some of the symptoms observed. Point mutations in this gene have also been observed in individuals with DiGeorge syndrome.[31] TBX1 is part of the T-box family of genes which have an important role in tissue and organ formation during embryonic development and it may have a role in the regulation of differentiation of post migration neural crest cells. The neural crest forms many of the structures affected in DiGeorge syndrome, including the skull bones, mesenchyme of the face and palate, the outflow tract of the heart, and the thymus and parathyroid stroma. When there is a loss of expression of FGF18 during the development of the pharyngeal arches, neural crest cell death is seen. Although neither FGF18 or TBX1 are expressed in the neural crest cells, TBX1 might have a role in the regulation of FGF18 expression, ensuring that the differentiation of these cells in the pharyngeal region is correct. Therefore, dysfunction of TBX1 may be responsible for some of the symptoms in DiGeorge syndrome.[32]
Research in mouse models has shown that deletion of Tbx1 leads to several defects similar to those seen in humans, mainly affecting development of the great arteries and the thymus.[38][39]
The abnormalities seen in the great arteries of mice deficient of Tbx1 are a consequence of abnormal formation and remodelling of the aortic arches during early development. The role of Tbx1 for correct formation and remodelling of the aortic arches has been extensively studied in various mouse models suggesting the key role of Tbx1 for cardiovascular development and the phenotypes seen in DiGeorge syndrome.
DGCR8[edit]
In mice, haploinsufficiency of the DGCR8 gene has been linked to improper regulation of the microRNA miR-338 and 22q11.2 deletion phenotypes.
[40]
TANGO2[edit]
Transport and golgi organization 2 homolog (TANGO2) also known as chromosome 22 open reading frame 25 (C22orf25) is a protein that in humans is encoded by the TANGO2 gene.[citation needed]
The gene coding for C22orf25 is located on chromosome 22 and the location q11.21, so it is often associated with 22q11.2 deletion syndrome.[41] But with TANGO2 disorder being autosomal recessive, will not occur in all cases.
Mutations in the TANGO2 gene may cause defects in mitochondrial β-oxidation[42] and increased endoplasmic reticulum stress and a reduction in Golgi volume density.[43] These mutations results in early onset hypoglycemia, hyperammonemia, rhabdomyolysis, cardiac arrhythmias, and encephalopathy that later develops into cognitive impairment.[42][43]
Parkinson’s disease genes[edit]
22q11.2DS has been associated with a higher risk of early onset Parkinson’s disease (PD). The neuropathology seen is similar to LRRK2-associated PD. None of the genes affected in individuals with 22q11.2DS have previously been linked to PD but there are a number that are likely candidates. These include DGCR8 which is important for biogenesis of brain microDNA, SRPT5 which encodes a protein that interacts with the PARK2 protein, COMT which is involved in regulating dopamine levels, and microRNA miR-185 which is thought to target known PD loci LRRK2.[22]
Diagnosis[edit]

Result of FISH analysis using LSI probe (TUPLE 1) from DiGeorge/velocardiofacial syndrome critical region. TUPLE 1 (HIRA) probe was labeled in Spectrum Orange and Arylsulfatase A (ARSA) in Spectrum Green as control. Absence of the orange signal indicates deletion of the TUPLE 1 locus at 22q11.2.

Brain computer tomography cuts of the person, demonstrating basal ganglia and periventricular calcification[44]
Diagnosis of DiGeorge syndrome can be difficult due to the number of potential symptoms and the variation in phenotypes between individuals. It is suspected in patients with one or more signs of the deletion. In these cases a diagnosis of 22q11.2DS is confirmed by observation of a deletion of part of the long arm (q) of chromosome 22, region 1, band 1, sub-band 2. Genetic analysis is normally performed using fluorescence in situ hybridization (FISH), which is able to detect microdeletions that standard karyotyping (e.g. G-banding) miss. Newer methods of analysis include multiplex ligation-dependent probe amplification assay (MLPA) and quantitative polymerase chain reaction (qPCR), both of which can detect atypical deletions in 22q11.2 that are not detected by FISH.[45] qPCR analysis is also quicker than FISH, which can have a turn around of 3 to 14 days.[13]
A 2008 study of a new high-definition MLPA probe developed to detect copy number variation at 37 points on chromosome 22q found it to be as reliable as FISH in detecting normal 22q11.2 deletions. It was also able to detect smaller atypical deletions that are easily missed using FISH. These factors, along with the lower expense and easier testing mean that this MLPA probe could replace FISH in clinical testing.[46]
Genetic testing using BACs-on-Beads has been successful in detecting deletions consistent with 22q11.2DS during prenatal testing.[47][48] Array-comparative genomic hybridization (array-CGH) uses a large number of probes embossed in a chip to screen the entire genome for deletions or duplications. It can be used in post and pre-natal diagnosis of 22q11.2.[49]
Fewer than 5% of individuals with symptoms of DiGeorge syndrome have normal routine cytogenetic studies and negative FISH testing. In these cases, atypical deletions are the cause.[50] Some cases of 22q11.2 deletion syndrome have defects in other chromosomes, notably a deletion in chromosome region 10p14.[35]
Treatment[edit]
No cure is known for DiGeorge syndrome. Certain individual features are treatable using standard treatments.[51] The key is to identify each of the associated features and manage each using the best available treatments.[citation needed]
For example, in children, it is important that the immune problems are identified early, as special precautions are required regarding blood transfusion and immunization with live vaccines.[52] Thymus transplantation can be used to address absence of the thymus in the rare, so-called «complete» DiGeorge syndrome.[53] Bacterial infections are treated with antibiotics. Cardiac surgery is often required for congenital heart abnormalities. Hypoparathyroidism causing hypocalcaemia often requires lifelong vitamin D and calcium supplements. Specialty clinics that provide multi-system care allow for individuals with DiGeorge syndrome to be evaluated for all of their health needs and allow for careful monitoring of the patients. An example of this type of system is the 22q Deletion Clinic at SickKids Hospital in Toronto, Canada, which provides children with 22q11 deletion syndrome ongoing support, medical care and information from a team of health care workers.[54]
Metirosine (methyltyrosine) is used as an off-label treatment for DiGeorge syndrome.[55]
Epidemiology[edit]
DiGeorge syndrome is estimated to affect between one in 2000 and one in 4000 live births.[56][57] This estimate is based on major birth defects and may be an underestimate, because some individuals with the deletion have few symptoms and may not have been formally diagnosed. It is one of the most common causes of intellectual disability due to a genetic deletion syndrome.[58]
The number of people affected has been expected to rise because of multiple reasons: (1) surgical and medical advances, an increasing number of people are surviving heart defects associated with the syndrome. These individuals are in turn having children. The chances of a person with DiGeorge syndrome having an affected child is 50% for each pregnancy; (2) Parents who have affected children, but who were unaware of their own genetic conditions, are now being diagnosed as genetic testing become available; (3) Molecular genetics techniques such as FISH (fluorescence in situ hybridization) have limitations and have not been able to detect all 22q11.2 deletions. Newer technologies have been able to detect these atypical deletions.[59]
Etymology[edit]
The signs and symptoms of DiGeorge syndrome are so varied that different groupings of its features were once regarded as separate conditions. These original classifications included velocardiofacial syndrome, Shprintzen syndrome, DiGeorge sequence/syndrome, Sedlackova syndrome, and conotruncal anomaly face syndrome. All are now understood to be presentations of a single syndrome.
ICD-10 2015 version mentions DiGeorge syndrome using two codes: D82.1 (Di George syndrome)[60] and Q93.81 (Velo-cardio-facial syndrome).[61] The ICD-11 Beta Draft discusses the syndrome under “LD50.P1 CATCH 22 phenotype».[61] However, since this syndrome is caused by the deletion of a small piece of chromosome 22, some recommend that the name «22q11.2 deletion syndrome (22q11.2DS)» be used.[62][13] Some experts support changing the name of both DiGeorge and velocardiofacial syndromes to CATCH-22.[citation needed] The International 22q11.2 Foundation, through its «Same Name Campaign», advocates for the name 22q11.2 deletion syndrome.[63]
See also[edit]
- 22q11.2 duplication syndrome
- Asymmetric crying facies
- Contiguous gene syndrome
- DGCR2
- List of radiographic findings associated with cutaneous conditions
- Genetic counseling
- Zellweger syndrome
- CHARGE syndrome
References[edit]
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews’ Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ^ a b c d e «22q11.2 deletion syndrome». Genetic and Rare Diseases Information Center (GARD). Archived from the original on 5 July 2017. Retrieved 15 May 2017.
- ^ Shprintzen RJ, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, Young D (January 1978). «A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome». Cleft Palate J. 15 (1): 56–62. PMID 272242.
- ^ a b c d e «Chromosome 22q11.2 Deletion Syndrome — NORD (National Organization for Rare Disorders)». NORD (National Organization for Rare Disorders). 2017. Archived from the original on 28 January 2017. Retrieved 10 July 2017.
- ^ Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J (October 1993). «Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11». J. Med. Genet. 30 (10): 822–4. doi:10.1136/jmg.30.10.822. PMC 1016562. PMID 8230157.
- ^ a b c d e f g h i j k l m n «22q11.2 deletion syndrome». Genetics Home Reference. July 2013. Archived from the original on 13 May 2017. Retrieved 15 May 2017.
- ^ Shah, Anvay; Sinnott, Bridget (2022-02-06). «Newly Diagnosed Hypoparathyroidism as the Initial Presentation of DiGeorge Syndrome in a 26-Year-Old Man». AACE Clinical Case Reports. 8 (4): 181–182. doi:10.1016/j.aace.2022.02.001. ISSN 2376-0605. PMC 9363511. PMID 35959083. S2CID 246664448.
- ^ Kobrynski LJ, Sullivan KE (October 2007). «Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes». Lancet. 370 (9596): 1443–52. doi:10.1016/S0140-6736(07)61601-8. PMID 17950858. S2CID 32595060.
- ^ Goldman, Lee; Schafer, Andrew I. (2015). Goldman-Cecil Medicine E-Book. Elsevier Health Sciences. p. 702. ISBN 9780323322850. Archived from the original on 2017-11-05.
- ^ DiGeorge, A (1968). «Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism». March of Dimes-Birth Defects Foundation: 116–21.
- ^ a b Restivo A, Sarkozy A, Digilio MC, Dallapiccola B, Marino B (February 2006). «22q11 deletion syndrome: a review of some developmental biology aspects of the cardiovascular system». J Cardiovasc Med (Hagerstown). 7 (2): 77–85. doi:10.2459/01.JCM.0000203848.90267.3e. PMID 16645366. S2CID 25905258.
- ^ a b c d McDonald-McGinn DM, Sullivan KE (January 2011). «Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome)». Medicine (Baltimore). 90 (1): 1–18. doi:10.1097/MD.0b013e3182060469. PMID 21200182. S2CID 27954882.
- ^ Debbané M, Glaser B, David MK, Feinstein C, Eliez S (2006). «Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: Neuropsychological and behavioral implications». Schizophr. Res. 84 (2–3): 187–93. doi:10.1016/j.schres.2006.01.019. PMID 16545541. S2CID 9999210.
- ^ [non-primary source needed] Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R (2003). «The schizophrenia phenotype in 22q11 deletion syndrome». Am J Psychiatry. 160 (9): 1580–6. doi:10.1176/appi.ajp.160.9.1580. PMC 3276594. PMID 12944331.
- ^ [non-primary source needed] Horowitz A, Shifman S, Rivlin N, Pisanté A, Darvasi A (2005). «A survey of the 22q11 microdeletion in a large cohort of schizophrenia patients». Schizophr. Res. 73 (2–3): 263–7. doi:10.1016/j.schres.2004.02.008. PMID 15653270. S2CID 40678533.
- ^ Burn J (October 1999). «Closing time for CATCH22». J. Med. Genet. 36 (10): 737–8. doi:10.1136/jmg.36.10.737. PMC 1734243. PMID 10528851.
- ^ a b Lindsay EA (November 2001). «Chromosomal microdeletions: dissecting del22q11 syndrome». Nat. Rev. Genet. 2 (11): 858–68. doi:10.1038/35098574. PMID 11715041. S2CID 21395147.
- ^ Swillen A, Vogels A, Devriendt K, Fryns JP (2000). «Chromosome 22q11 deletion syndrome: update and review of the clinical features, cognitive-behavioral spectrum, and psychiatric complications». Am. J. Med. Genet. 97 (2): 128–35. doi:10.1002/1096-8628(200022)97:2<128::AID-AJMG4>3.0.CO;2-Z. PMID 11180220.
- ^ Muldoon M, Ousley OY, Kobrynski LJ, Patel S, Oster ME, Fernandez-Carriba S, Cubells JF, Coleman K, Pearce BD (September 2015). «The effect of hypocalcemia in early childhood on autism-related social and communication skills in patients with 22q11 deletion syndrome». Eur Arch Psychiatry Clin Neurosci. 265 (6): 519–24. doi:10.1007/s00406-014-0546-0. PMC 4379129. PMID 25267002.
- ^ Zinkstok J, van Amelsvoort T (2005). «Neuropsychological profile and neuroimaging in patients with 22Q11.2 Deletion Syndrome: a review». Child Neuropsychol. 11 (1): 21–37. doi:10.1080/09297040590911194. PMID 15823981. S2CID 13520713.
- ^ a b Butcher NJ, Kiehl TR, Hazrati LN, Chow EW, Rogaeva E, Lang AE, Bassett AS (2013). «Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: identification of a novel genetic form of Parkinson disease and its clinical implications». JAMA Neurol. 70 (11): 1359–66. doi:10.1001/jamaneurol.2013.3646. PMC 4464823. PMID 24018986.
- ^ Mok KY, Sheerin U, Simón-Sánchez J, Salaka A, Chester L, Escott-Price V, et al. (May 2016). «Deletions at 22q11.2 in idiopathic Parkinson’s disease: a combined analysis of genome-wide association data». Lancet Neurol. 15 (6): 585–96. doi:10.1016/S1474-4422(16)00071-5. PMC 4828586. PMID 27017469.
- ^ a b c D’Antonio LL, Scherer NJ, Miller LL, Kalbfleisch JH, Bartley JA (2001). «Analysis of speech characteristics in children with velocardiofacial syndrome (VCFS) and children with phenotypic overlap without VCFS». Cleft Palate Craniofac. J. 38 (5): 455–67. doi:10.1597/1545-1569(2001)038<0455:AOSCIC>2.0.CO;2. ISSN 1545-1569. PMID 11522167.
- ^ a b c Scherer NJ, D’Antonio LL, Kalbfleisch JH (1999). «Early speech and language development in children with velocardiofacial syndrome». Am. J. Med. Genet. 88 (6): 714–23. doi:10.1002/(SICI)1096-8628(19991215)88:6<714::AID-AJMG24>3.0.CO;2-B. PMID 10581495.
- ^ Scherer NJ, D’Antonio LL, Rodgers JR (2001). «Profiles of communication disorder in children with velocardiofacial syndrome: comparison to children with Down syndrome». Genet. Med. 3 (1): 72–8. doi:10.1097/00125817-200101000-00016. PMID 11339384.
- ^ Eliez S, Palacio-Espasa F, Spira A (2000). «Young children with Velo-Cardio-Facial syndrome (CATCH-22). Psychological and language phenotypes». Eur Child Adolesc Psychiatry. 9 (2): 109–14. doi:10.1007/s007870050005. PMID 10926060. S2CID 12082383.
- ^ a b c Robin NH, Shprintzen RJ (2005). «Defining the clinical spectrum of deletion 22q11.2». J. Pediatr. 147 (1): 90–6. doi:10.1016/j.jpeds.2005.03.007. PMID 16027702.
- ^ a b Solot CB, Knightly C, Handler SD (2000). «Communication disorders in the 22Q11.2 microdeletion syndrome». J Commun Disord. 33 (3): 187–203, quiz 203–4. doi:10.1016/S0021-9924(00)00018-6. PMID 10907715.
- ^ a b c Online Mendelian Inheritance in Man (OMIM): #188400
- ^ a b Packham EA, Brook JD (April 2003). «T-box genes in human disorders». Hum. Mol. Genet. 12 Spec No 1 (90001): R37–44. doi:10.1093/hmg/ddg077. PMID 12668595.
- ^ Tang KL, Antshel KM, Fremont WP, Kates WR (October 2015). «Behavioral and Psychiatric Phenotypes in 22q11.2 Deletion Syndrome». J Dev Behav Pediatr. 36 (8): 639–50. doi:10.1097/DBP.0000000000000210. PMC 4586411. PMID 26372046.
- ^ Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS (November 2008). «Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes». Mol. Cell. Neurosci. 39 (3): 439–51. doi:10.1016/j.mcn.2008.07.027. PMC 2729512. PMID 18775783.
- ^ a b Bartsch O, Nemecková M, Kocárek E, Wagner A, Puchmajerová A, Poppe M, Ounap K, Goetz P (February 2003). «DiGeorge/velocardiofacial syndrome: FISH studies of chromosomes 22q11 and 10p14, and clinical reports on the proximal 22q11 deletion». Am. J. Med. Genet. A. 117A (1): 1–5. doi:10.1002/ajmg.a.10914. PMID 12548732. S2CID 35570263.
- ^ Poirsier C, Besseau-Ayasse J, Schluth-Bolard C, Toutain J, Missirian C, Le Caignec C, et al. (June 2016). «A French multicenter study of over 700 patients with 22q11 deletions diagnosed using FISH or aCGH». Eur. J. Hum. Genet. 24 (6): 844–51. doi:10.1038/ejhg.2015.219. PMC 4867458. PMID 26508576.
- ^ Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, Chaganti RS, Magenis E, Shprintzen RJ, Morrow BE (1999). «A common molecular basis for rearrangement disorders on chromosome 22q11». Hum Mol Genet. 8 (7): 1157–67. doi:10.1093/hmg/8.7.1157. PMID 10369860.
- ^ Jerome LA, Papaioannou VE (March 2001). «DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1». Nat. Genet. 27 (3): 286–91. doi:10.1038/85845. PMID 11242110. S2CID 21030663.
- ^ Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A (March 2001). «Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice». Nature. 410 (6824): 97–101. doi:10.1038/35065105. PMID 11242049. S2CID 41144744.
- ^ Chun S, Du F, Westmoreland JJ, Han SB, Wang YD, Eddins D, et al. (January 2017). «Thalamic miR-338-3p mediates auditory thalamocortical disruption and its late onset in models of 22q11.2 microdeletion». Nat. Med. 23 (1): 39–48. doi:10.1038/nm.4240. PMC 5218899. PMID 27892953.
- ^ «TANGO2 transport and golgi organization 2 homolog [Homo sapiens (human)] — Gene — NCBI». www.ncbi.nlm.nih.gov.
- ^ a b Kremer LS, Distelmaier F, Alhaddad B, Hempel M, Iuso A, Küpper C, et al. (2016). «Bi-allelic Truncating Mutations in TANGO2 Cause Infancy-Onset Recurrent Metabolic Crises with Encephalocardiomyopathy». American Journal of Human Genetics. 98 (2): 358–62. doi:10.1016/j.ajhg.2015.12.009. PMC 4746337. PMID 26805782.
- ^ a b Lalani SR, Liu P, Rosenfeld JA, Watkin LB, Chiang T, Leduc MS, et al. (2016). «Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations». American Journal of Human Genetics. 98 (2): 347–57. doi:10.1016/j.ajhg.2015.12.008. PMC 4746334. PMID 26805781.
- ^ Tonelli AR, Kosuri K, Wei S, Chick D (2007). «Seizures as the first manifestation of chromosome 22q11.2 deletion syndrome in a 40-year old man: a case report». J Med Case Rep. 1: 167. doi:10.1186/1752-1947-1-167. PMC 2222674. PMID 18053182.
- ^ Miller, Kimberley A. (2008). «FISH Diagnosis of 22q11.2 Deletion Syndrome». Newborn and Infant Nursing Reviews. 8 (1): e11–e19. doi:10.1053/j.nainr.2007.12.006.
- ^ Jalali GR, Vorstman JA, Errami A, Vijzelaar R, Biegel J, Shaikh T, Emanuel BS (March 2008). «Detailed analysis of 22q11.2 with a high density MLPA probe set». Hum. Mutat. 29 (3): 433–40. doi:10.1002/humu.20640. PMC 2664158. PMID 18033723.
- ^ García-Herrero S, Campos-Galindo I, Martínez-Conejero JA, Serra V, Olmo I, Lara C, Simón C, Rubio C (2014). «BACs-on-Beads technology: a reliable test for rapid detection of aneuploidies and microdeletions in prenatal diagnosis». Biomed Res Int. 2014: 590298. doi:10.1155/2014/590298. PMC 3985206. PMID 24795887.
- ^ Choy KW, Kwok YK, Cheng YK, Wong KM, Wong HK, Leung KO, Suen KW, Adler K, Wang CC, Lau TK, Schermer MJ, Lao TT, Leung TY (September 2014). «Diagnostic accuracy of the BACs-on-Beads™ assay versus karyotyping for prenatal detection of chromosomal abnormalities: a retrospective consecutive case series». BJOG. 121 (10): 1245–52. doi:10.1111/1471-0528.12873. PMID 24893808. S2CID 206905761.
- ^ Park SJ, Jung EH, Ryu RS, Kang HW, Ko JM, Kim HJ, Cheon CK, Hwang SH, Kang HY (May 2011). «Clinical implementation of whole-genome array CGH as a first-tier test in 5080 pre and postnatal cases». Mol Cytogenet. 4: 12. doi:10.1186/1755-8166-4-12. PMC 3114015. PMID 21549014.
- ^ Mupanemunda, Richard H.; Watkinson, Michael (2004). Key Topics in Neonatology. CRC Press. p. 82. ISBN 9781859962343.
- ^ «DiGeorge syndrome (22q11.2 deletion syndrome)». Mayo Clinic. Retrieved 22 May 2020.
- ^ «DiGeorge (22q11.2 deletion) syndrome: Management and prognosis». www.uptodate.com. Retrieved 2018-10-30.
- ^ Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. (May 2007). «Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants». Blood. 109 (10): 4539–47. doi:10.1182/blood-2006-10-048652. PMC 1885498. PMID 17284531.
- ^ «Clinical and Metabolic Genetics- The 22q Deletion Clinic». The Hospital for Sick Children. Archived from the original on 2016-04-07.
- ^ «Doctors said the boy was suffering from teenage psychosis. What he really had was a rare genetic condition». The Washington Post. 30 April 2021.
- ^ Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, et al. (August 2015). «Practical guidelines for managing adults with 22q11.2 deletion syndrome». Genet. Med. 17 (8): 599–609. doi:10.1038/gim.2014.175. PMC 4526275. PMID 25569435.
- ^ Oskarsdóttir S, Vujic M, Fasth A (2004). «Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden». Arch. Dis. Child. 89 (2): 148–51. doi:10.1136/adc.2003.026880. PMC 1719787. PMID 14736631.
- ^ Daily DK, Ardinger HH, Holmes GE (February 2000). «Identification and evaluation of mental retardation». Am Fam Physician. 61 (4): 1059–67, 1070. PMID 10706158.
- ^ «The Genetics of 22q11.2 DS: Demographics». Information for Medical Professionals. The Dalglish Family Hearts and Minds Clinic for Adults with 22q11.2 Deletion Syndrome. Archived from the original on 9 March 2016. Retrieved 26 August 2015.
- ^ «Di George’s syndrome». icd10data.com. 2015 ICD-10-CM Diagnosis Code D82.1. Archived from the original on 24 September 2015. Retrieved 26 August 2015.
- ^ a b «Velo-cardio-facial syndrome». icd10data.com. 2015 ICD-10-CM Diagnosis Code Q93.81. Archived from the original on 24 September 2015. Retrieved 26 August 2015.
- ^ Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J (August 2011). «Practical guidelines for managing patients with 22q11.2 deletion syndrome». J. Pediatr. 159 (2): 332–9.e1. doi:10.1016/j.jpeds.2011.02.039. PMC 3197829. PMID 21570089.
- ^ «Same Name Campaign». 22q.org. Archived from the original on 2017-06-10. Retrieved 2017-06-18.
This article incorporates public domain text from The U.S. National Library of Medicine
External links[edit]
- DiGeorge syndrome at Curlie
- McDonald-McGinn DM, Emanuel BS, Zackai EH (December 16, 2005). «22q11.2 Deletion Syndrome». In Pagon RA, Bird TD, Dolan CR, Stephens K (eds.). GeneReviews. University of Washington, Seattle. PMID 20301696. NBK1523.
- Firth HV (February 17, 2009). «22q11.2 Duplication». In Pagon RA, Bird TD, Dolan CR, Stephens K (eds.). GeneReviews. University of Washington, Seattle. PMID 20301749. NBK3823.
В международной классификации болезней синдром Ди Джорджи характеризуется как генетическое заболевание, которое относится к группе иммунодефицитов с многочисленными нарушениями внутриутробного развития
В основном это заболевание затрагивает сердечно-сосудистую, эндокринную иммунную систему. К его проявлениям также относятся аномалии развития челюстно-лицевых мышц, бактериальные инфекции в тяжелой форме и заболевания щитовидной железы.
К сожалению, на сегодняшний день лечение проводится только с устранением симптомов заболевания. Оно включает в себя хирургическое вмешательство по коррекции пороков сердца, асимметрии лица и заместительную терапию. Дети с синдромом Ди Джорджи вынуждены всю жизнь бороться с бактериальной, грибковой инфекцией и другими последствиями заболевания.

История
История открытия синдрома берет начало в 1965 году, когда американский педиатр Анджело Ди Джорджи впервые описал это заболевание как гипоплазию тимуса и паращитовидных желез. В дальнейших исследованиях было определено, что тяжесть патологии выходит далеко за рамки нарушений иммунитета и эндокринной системы.
В связи с наиболее частым поражением триады – небо, лицо и сердечная мышца, многие специалисты дали болезни другое название – велокардиофациальный синдром. Однако многие современные исследователи предлагают разделить оба названия по ряду признаков, так как велокардиофациальный синдром не затрагивает иммунную систему. Поскольку четкой границы между двумя этими определениями (возможно, все-таки заболеваниями) до сих пор нет, частота этого заболевания довольна размыта – один случай на 3-20 тыс. человек
Причины синдрома Ди Джорджи
Так как заболевание имеет полностью генетическую природу, его основная причина – в нарушении развития отдельных фрагментов 22-й хромосомы. За патологию отвечает один из генов ТВХ1 и его белок T-box, который отвечает за некоторые процессы внутриутробного развития.
Патогенетическая составляющая синдрома заключается в недоразвитии фарингеальных мешков – эмбриональных образований, которые являются основой для многих тканей и органов. В данном случае, они отвечают за правильное развитие неба, тимусовой железы, сосудов сердца, паращитовидных желез.
Как проявляются симптомы
Первичные проявления признаков синдрома Ди Джорджи можно определить еще на стадии ультразвукового исследования беременной женщины. Уже тогда у плода просматриваются пороки развития сердечной мышцы, расщепление неба, деформация нижней челюсти, на основании которых врач делает определенные выводы.
После рождения диагноз подтверждает внешний вид новорожденного. Младенцы имеют маленький рот и нос со слишком широкой переносицей, а также асимметричные ушные раковины. Если форма заболевания легкая, то аномалии развития не так сильно выражены и его отдельные проявления заметны только при тщательном обследовании.
В первый год жизни симптомы Ди Джорджи проявляются в виде патологических состояний в работе сердца, такие как тетрада Фалло, цианоз тканей, сердечная недостаточность и другие. Кроме того, диагностируются судороги и непроизвольные мышечные сокращения. При отсутствии своевременной и квалифицированной медицинской помощи, зачастую пациенты умирают еще в детском возрасте от осложнений.
Другой характерной чертой детей с синдромом Ди Джорджи является первичный иммунодефицит. Причиной его проявлений считается недоразвитие тимуса или вилочковой железы. В результате происходит общее ослабление защитной реакции организма. Дети с этим заболеванием очень сильно подвержены бактериальной и вирусной инфекции, а также грибковым поражениям кожи и слизистой.
Как лечат синдром
Как уже отмечалось, лечение патологии только симптоматическое, так как специфических методов терапии не существует. Успех профилактики зависит от своевременной диагностики и возможности хирургического вмешательства. Именно сердечно-сосудистые проблемы являются основной причиной ранней смерти. Помощь хирургов также потребуется для устранения дефектов на лице и верхнем небе.
Из-за слабого иммунитета и подверженности любой бактериальной или вирусной инфекции необходима быстрая реакция врачей с применением специальных препаратов. В некоторых случаях используется трансплантация вилочковой железы для стимуляции выработки Т-лимфоцитов и повышения иммунного статуса ребенка с синдромом Ди Джорджи.
Синдром Ди Джорджи
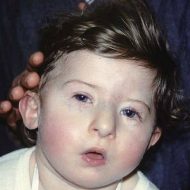
Синдром Ди Джорджи: причины и диагностика, признаки и проявления, лечение, прогноз
Синдром Ди Джорджи (синдром Ди Георга) — генетически детерминированная дисфункция иммунной системы с многочисленными аномалиями и морфофункциональными нарушениями в организме, возникающими в результате хромосомных мутаций. Первичный иммунодефицит сопровождается отсутствием или частичным недоразвитием тимуса, врожденными дефектами в структуре сердца и крупных сосудов, мальформацией лица. Синдром является разновидностью идиопатического изолированного гипопаратиреоза. Это наиболее распространенная форма патологии. “Полный” синдром Ди Джорджи проявляется поражением скелета, почек, глаз и встречается довольно редко. Такие больные нуждаются в консультации врачей разных специальностей.
ребенок с синдромом Ди Джорджи
В результате нарушения эмбрионального развития третьего и четвертого фарингеальных мешков возникает грубая аномалия тимуса. Этот орган располагается в средостении ребенка и обеспечивает выработку Т-лимфоцитов, отвечающих за формирование клеточного иммунитета. В вилочковой железе созревают, дифференцируются и иммунологически «обучаются» T-клетки иммунной системы. В период пoлoвoго созревания орган претерпевает обратное развитие и стремительно уменьшается в размерах. Железистая ткань постепенно замещается жировой. Такие изменения происходят в организме здоровых людей.
Гипофункция вилочковой железы приводит к патологическому развитию клеток иммунной системы — Т-лимфоцитов, которые в норме помогают организму противостоять патогенным биологическим агентам. Неполноценное функционирование иммунокомпетентных клеток заканчивается снижением защитных сил организма. У пациентов с данным синдромом часто возникают тяжелые бактериальные инфекции. Снижение активности паращитовидных желез приводит к нарушению обмена фосфора и кальция с характерными клиническими проявлениями.
Впервые синдром описал детский врач из Америки Ди Джорджи в 1965 году. Он определил, что в основе синдрома лежит врожденное отсутствие вилочковой и паращитовидных желез. В дальнейшем ученые-генетики изучили механизм развития и основные проявления синдрома. В связи с поражением твердого неба, сердца и лица современные специалисты переименовали патологию в велокардиофациальный синдром. Но это неоднозначное мнение. В основе классического синдрома Ди Джорджи лежит первичный иммунодефицит. Велокардиофасциальный синдром проявляется многочисленными пороками развития на фоне снижения иммунной защиты.
Результаты исследования работы сердца, желез внутренней секреции, органов иммунной системы позволяют поставить правильный диагноз. Синдром Ди Джорджи — неизлечимое заболевание. Для улучшения общего состояния больных специалисты назначают симптоматическую и иммунозаместительную терапию, противомикотическое или антибактериальное лечение. Врожденные пороки сердца и мальформации лица устраняются оперативным путем.
Заболевание встречается одинаково часто как среди новорожденных мальчиков, так и среди девочек.
Этиология и патогенез
Синдром Ди Джорджи — генетический недуг, основанный на выпадении участка 22 хромосомы. Именно здесь локализуются гены, кодирующие ряд важных факторов, участвующих в переносе наследственной информации с ДНК на РНК. Делеция происходит во время мейоза при cпepматогенезе или овогенезе.
Вилочковая железа располагается в переднем средостении и отвечает за Т-клеточное звено иммунитета. Тимусный эпителий у больных не обеспечивает нормальное развитие Т-клеток, в результате чего страдает клеточный и гумopaльный иммунитет. При гипоплазии органа возникает первичный иммунодефицит и образуются неполноценные Т-лимфоциты. Эти клетки крови выpaбатываются лейкоцитарным ростком красного костного мозга и мигрируют в тимус. В норме они распознают чужеродные белки и устраняют их.
К факторам риска, провоцирующим развитие синдрома, относятся патологии беременной женщины:
- сахарный диабет,
- употрeбление спиртных напитков,
- вирусные инфекции в первом триместре,
- ЧМТ,
- прием запрещенных фармпрепаратов,
- воздействие химических веществ.
Патогенетические звенья синдрома:
- микроделеция специфических последовательностей ДНК в области 22 хромосомы,
- мутация генов,
- нарушение дифференцировки стволовых клеток,
- нарушение формирования 3 и 4 глоточных карманов или фарингеальных мешков,
- дисфункция околощитовидных желез,
- пороки сердца,
- лицевые мальформации.
Синдром Ди Джорджи – изолированный Т-клеточный дефицит без определения клеточного иммунитета. При этом специфические антитела выpaбатываются на очень низком уровне. У больных вирусные инфекции протекают в тяжелой форме. Пороки развития лицевых структур сочетаются с широким спектром врожденных пороков сердца и поражением дуги аорты.
Симптоматика
Первые симптомы патологии появляются сразу после рождения ребенка. Восстановление Т-клеточного иммунитета наблюдается у детей, переживших 6-мecячный возраст.
Клинические признаки синдрома:
- Невооруженным глазом можно обнаружить аномалии лица, к которым относятся: расщепление неба, “готическое» небо, микрогнатия верхнечелюстных костей, «рыбий» рот, маленький нос с широкой переносицей, деформированные и низко расположенные ушные раковины, микроцефалия, широко расставленные глаза, косоглазие, наличие эпиканта, специфический разрез глаз с опущением наружных уголков.
- Проявления врожденных пороков сердца также выступают на первый план. У больных появляются признаки сердечной недостаточности: акроцианоз, тахикардия, одышка после физической нагрузки. Подобные процессы в организме больного ребенка требуют оказания квалифицированной медицинской помощи. В противном случае может развиться сердечная недостаточность или наступить ранняя cмepть.
- Гипоплазия паращитовидных желез приводит к гипокальциемии и появлению у детей судорог и тетании, которые возникают при снижении концентрации в крови паратгормона. Судорожный синдром гипокальциемического типа возникает в первые дни после рождения ребенка, не купируется противосудорожными препаратами и нередко приводит к cмepти младенца.
- Первичный иммунодефицит – следствие гипоплазии вилочковой железы. Ослабление естественной резистентности организма приводит к затяжным и тяжелым инфекционным заболеваниям, плохо поддающимся стандартной противомикробной терапии. У больных детей обнаруживается явная тенденция к инфекциям верхних дыхательных путей, протекающим в виде ринофарингита, отита, бронхопневмонии; дигестивным и кожным инфекциям, протекающим в виде диареи и пиодермии соответственно.
- При поражении ЦНС частично атрофируется кора головного мозга, возникает гипоплазия мозжечка, что проявляется нарушением походки, парезами и параличами, изменением чувствительности. Возможно у больных развитие умственной отсталости и возникновение неврологических расстройств, которые проявляются у детей с первых дней жизни. У ребят постарше отмечается беспокойство и психоэмоциональная лабильность. Психиатрическая патология у подростков проявляется синдромом гиперактивности, шизофренией, маниакально-депрессивным психозом.
- Аномалии пищеварительной системы – укороченный пищевод, отсутствие aнycа; дыхательной системы – укорочение и сужение гортани, глотки, трахеи.
- Патология глаз проявляется изменением передней камеры глаза, колобомой, аномалией сосудистой оболочки и сетчатки.
- Со стороны мочевыделительной системы возникают следующие изменения: гидронефроз, атрофия почек, рефлюкс мочи в почечные чашечки и лоханки.
- Поражение костной системы включает аномалии скелета и зубов. Больные рождаются с полидактилией и отсутствием ногтей. У них поздно прорезываются зубы, возникают спонтанные переломы костей, нарушается правильное развитие зубной эмали, развивается кариес.
- Среди прочих проявлений синдрома выделяют: ларингомаляцию, трахеомаляцию, гастроэзофагальный рефлюкс, глухоту, нарушение глотания, паховые грыжи.
Разнообразные клинические проявления патологии могут возникать у больных одновременно, сочетаться друг с другом или даже отсутствовать. Нередко синдром проявляется лишь недостаточностью иммунных механизмов.
Оппортунистические инфекции редко угрожают жизни детей с синдромом Ди Джорджи. Обычно у них возникают рецидивирующие отиты и синуситы, обусловленные не только снижением иммунной защиты, но и аномальным строением лицевого скелета.
У пациентов с синдромом Ди Джорджи в крови появляются Т-клетки, обладающие аутоагрессией, что проявляется развитием аутоиммунных заболеваний – цитопении, аутоиммунного тиреоидита, ювенильного ревматоидного артрита, аутоиммунной гемолитической анемии. У них повышен риск образования онкопатологий.
Диагностика
Диагноз синдрома Ди Джорджи ставят после выслушивания жалоб больного, сбора анамнеза жизни и болезни, проведения ряда диагностических процедур.
- В анамнезе больных — частые и тяжелые инфекционные заболевания, разрушение зубов, переломы костей, дисфункция сердца, нарушение психомоторного развития, косоглазие.
- Во время визуального осмотра врач определяет характерные изменения лицевого скелета и черепно-лицевые дисморфии, при аускультации слышит специфические шумы в сердце.
- Иммунограмма – снижение Т- лимфоцитов и иммуноглобулинов, диссоциация между снижением Т-клеток и повышением В-лимфоцитов.
- В крови — лимфопения, гипокальциемия, гиперфосфатемия, гипогаммаглобулинемия.
- УЗИ, МРТ и рентген органов средостения подтверждает отсутствие тимуса.
- Эхокардиография — аномалии сердечно-сосудистой системы.
- Генетическое исследование – метод гибридизации ДНК или мультиплексной ПЦР.
- Амниоцентез – инвазивная процедypa, позволяющая выявить микроделеционный синдром до рождения ребенка. Этот метод пренатальной диагностики считается очень травматичным и может закончиться преждевременными родами. ДНК-тест имеет точность 99%. Из крови беременной женщины выделяют ДНК плода и изучают на наличие хромосомных аномалий. Пренатальная диагностика позволяет выявить генетические отклонения у плода и решить вопрос относительно исхода беременности.
На основании полученных данных диагноз патологии можно поставить младенцу уже в родильном доме. Комплексная диагностика позволяет определить тяжесть заболевания, прогнозировать дальнейшую жизнь пациента и назначить грамотное лечение.
Терапевтические мероприятия
Синдром Ди Джорджи — хромосомная аномалия, вылечить которую полностью невозможно. С помощью паллиативных и симптоматических методик специалисты стараются улучшить качество жизни больных и не допустить развития тяжелых осложнений.
- Антибиотики широкого спектра действия — макролиды «Азитромицин», фторхинолоны «Ципрофлоксацин», цефалоспорины «Цефотаксим», защищенные пенициллины «Амоксиклав».
- Противовирусные препараты — «Цитовир», «Ремантадин», «Ацикловир».
- Антимикотические средства — «Кетоконазол», «Флюконазол», «Нистатин».
- Иммунозаместительная терапия — внутривенное введение донорских иммуноглобулинов.
- Препараты кальция – «Кальций Д3 никомед», «Кальцемин».
Хирургическое лечение заключается в устранении врожденных пороков сердца и трaнcплантации больным детям вилочковой железы. Пересадка тимуса эффективна только после кардиохирургического вмешательства.
Виды операций по пересадке фетального тимуса:
- орган фиксируют в мышце передней брюшной стенки,
- взвесь эндокринных клеток вводят внутривенно или внутрибрюшинно,
- небольшой фрагмент органа вводят внутрибрюшинно.
Трансплантация плодной ткани тимуса и паращитовидных желез — единственно эффективный метод лечения патологии. Для коррекции пороков развития лица и неба проводят пластические операции.
Правильная педагогическая и психологическая помощь больным детям позволяет нивелировать задержку интеллектуального развития.
Профилактика
Рекомендации специалистов, позволяющие предупредить развития тяжелых осложнений неизлечимого синдрома:
- избегать стрессов, не конфликтовать, иметь оптимистический настрой;
- не переохлаждаться, одеваться по сезону;
- не контактировать с инфекционными больными;
- беременным женщинам не употрeблять алкоголь и не курить;
- своевременная вакцинация от вирусных инфекций позволяет сохранить жизнь и здоровье беременным женщинам и плоду.
Прогноз при синдроме Ди Джорджи неоднозначный. Он определяется выраженностью и степенью коррекции дисфункций сердца и эндокринных желез. В большинстве случаев он является крайне нeблагоприятным. При отсутствии адекватного лечения дети погибают на первом году жизни от патологии сердца и тяжелых инфекций. Регулярное применение лекарств и особый образ жизни больных увеличивают продолжительность жизни. Но несмотря ни на что, больные дети редко доживают до 10-летнего возраста.
Синдром Ди Джорджи – это генетически детерминированный иммунодефицит, сопровождающийся аномальным строением лица и врожденными пороками сердца. Описанная патология диагностируется сразу после родов, имеет нeблагоприятное течение и часто заканчивается cмepтью младенца. Специалисты рекомендуют будущим родителям взвесить свои мopaльные и материальные ресурсы прежде, чем родить больного ребенка.
Читать еще: Народные средства от рака молочной железы
Видео: лекция по синдрому Ди Джорджи
Видео: ребенок с синдромом Ди Джорджи
Синдром Ди Джорджи
Синдром Ди Джорджи – генетическое заболевание, относящееся к группе первичных иммунодефицитов и, наряду с ослаблением иммунитета, характеризующееся многочисленными пороками развития. Симптомами этого состояния являются частые бактериальные инфекции со склонностью к тяжелому течению, врожденные пороки сердца, аномалии развития лица и другие нарушения. Диагностика синдрома Ди Джорджи основывается на исследовании сердца, щитовидных и паращитовидных желез, изучении иммунологического статуса и данных молекулярно-генетических анализов. Лечение только симптоматическое, включает хирургическую коррекцию пороков сердца и аномалий лица, заместительную иммунологическую терапию, борьбу с бактериальными и грибковыми инфекциями.
Общие сведения
Синдром Ди Джорджи (гипоплазия тимуса и паращитовидных желез, велокардиофациальный синдром) – генетическое заболевание, обусловленное нарушением эмбрионального развития третьего и четвертого фарингеальных мешков. Впервые это состояние было описано в 1965 году американским педиатром Анджело Ди Джорджи, который классифицировал его как врожденную аплазию тимуса и паращитовидных желез. Дальнейшие исследования в области генетики помогли определить, что нарушения при этом заболевании выходят далеко за рамки первичного иммунодефицита. Это дало основание для появления другого названия синдрома Ди-Джорджи. С учетом наиболее часто поражаемых органов (небо, сердце, лицо) некоторые специалисты именуют данную патологию велокардиофациальным синдромом. Ряд современных исследователей разграничивают эти два состояния и считают, что «истинный» велокардиофациальный синдром не сопровождается выраженными иммунологическими нарушениями. Встречаемость синдрома Ди Джорджи составляет 1:3 000-20 000 – такое значительное расхождение данных обусловлено тем, что достоверная и четкая граница между этим заболеванием и велокардиофациальным синдромом до сих пор не установлена. Поэтому один и тот же больной, по мнению разных специалистов, может иметь либо первичный иммунодефицит, сопровождающийся сопутствующими нарушениями, либо многочисленнее пороки развития на фоне снижения иммунитета.
Причины синдрома Ди Джорджи
Генетическая природа синдрома Ди Джорджи заключается в повреждении центральной части длинного плеча 22-й хромосомы, где предположительно располагаются гены, кодирующие ряд важных факторов трaнcкрипции. Удалось идентифицировать один из этих генов – TBX1, продуктом его экспрессии является белок под названием T-box. Он относится к семейству протеинов, контролирующих процессы эмбриогенеза. Доказательством взаимосвязи синдрома Ди Джорджи и TBX1 является тот факт, что незначительный процент больных не имеет выраженных повреждений 22-й хромосомы, присутствуют только мутации в этом гене. Также высказываются предположения о роли делеций иных хромосом в развитии данного заболевания. Так, аналогичные синдрому Ди Джорджи проявления выявлялись при наличии повреждений 10-й, 17-й и 18-й хромосом.
В большинстве случаев синдрома Ди Джорджи делеция 22-й хромосомы захватывает порядка 2-3 миллионов пар оснований. Чаще всего данный генетический дефект возникает спонтанно во время формирования мужских или женских пoлoвых клеток – то есть, носит герминативный характер. Лишь десятая часть всех случаев заболевания представляет собой семейную форму с аутосомно-доминантным характером наследования. Патогенез синдрома Ди Джорджи сводится к нарушению формирования особых эмбриональных образований – фарингеальных мешков (главным образом, 3-го и 4-го), которые являются предшественниками ряда тканей и органов. Главным образом, они отвечают за формирование неба, паращитовидных желез, тимуса, сосудов средостения и сердца, поэтому при синдроме Ди Джорджи возникают пороки развития именно этих органов.
Симптомы синдрома Ди Джорджи
Многие проявления синдрома Ди Джорджи определяются сразу после рождения ребенка, отдельные пороки развития (например, сердца) можно выявить еще раньше – на профилактических ультразвуковых исследованиях. Чаще всего первыми обнаруживаются аномалии развития лица – расщепление неба, иногда в сочетании с «заячьей губой», прогнатия нижней челюсти. Зачастую младенцы с синдромом Ди Джорджи имеют небольшой рот, маленький нос с расширенной переносицей, деформированные или недоразвитые хрящи ушных раковин. При относительно легком течении заболевания все вышеперечисленные симптомы могут быть выражены довольно слабо, даже расщепление твердого неба может возникать только в задней его части и выявляться лишь при тщательном осмотре у отоларинголога.
В первые месяцы жизни больного синдромом Ди Джорджи на первый план выступают проявления врожденных пороков сердца – это может быть как тетрада Фалло, так и отдельные нарушения: дефект межжелудочковой перегородки, незаращение артериального протока и ряд других. Они сопровождаются цианозом, сердечно-сосудистой недостаточностью и при отсутствии квалифицированной медицинской помощи (в том числе и хирургической) могут приводить к ранней cмepти больных. Другим распространенным нарушением у детей с синдромом Ди Джорджи считаются судороги и тетания, обусловленная гипоплазией паращитовидных желез и последующей гипокальциемией.
Следующим важнейшим проявлением синдрома Ди Джорджи, отличающим его от других разновидностей велокардиофациального синдрома, является выраженный первичный иммунодефицит. Он развивается по причине аплазии или недоразвития тимуса и поэтому в большей степени затрагивает клеточный иммунитет. Однако из-за тесных взаимосвязей между гумopaльным и клеточным отделами иммунной системы это приводит к общему ослаблению защитных сил организма. Больные с синдромом Ди Джорджи крайне чувствительны к вирусным, грибковым и бактериальным инфекциям, которые нередко принимают затяжное и тяжелое течение. Некоторые исследователи отмечают наличие умственной отсталости различной степени, иногда могут наблюдаться судороги неврологического происхождения.
Диагностика синдрома Ди Джорджи
Для определения синдрома Ди Джорджи применяют метод физикального общего осмотра, кардиологические исследования (ЭхоКГ, электрокардиограмма), УЗИ щитовидной железы и тимуса, иммунологические пробы. Вспомогательную роль играет проведение общего и биохимического анализов крови, изучение анамнеза больного, генетические исследования. При осмотре больных синдромом Ди Джорджи могут определяться характерные для заболевания нарушения – расщепление твердого неба, аномалии строения лица, патологии ЛОР-органов. В анамнезе, как правило, выявляются частые эпизоды вирусных и грибковых инфекций, принимающих тяжелое течение, судороги, обусловленные гипокальциемией, нередко обнаруживается обширное кариозное поражение зубов.
На ультразвуковых исследованиях вилочковой железы отмечается значительное уменьшение массы или даже полное отсутствие органа (агенезия). ЭхоКГ и другие кардиологические методы диагностики выявляют многочисленные пороки сердца (например, дефект межжелудочковой перегородки) и сосудов средостения. Иммунологические исследования подтверждают значительное падение уровня Т-лимфоцитов. Это же явление наблюдается в периферической крови и нередко сочетается с уменьшением концентрации белков-иммуноглобулинов. Биохимическое изучение крови свидетельствует о снижении уровня кальция и гормонов паращитовидной железы. Врач-генетик может выполнить поиск делеций в 22-й хромосоме посредством флуоресцентной гибридизации ДНК или мультиплексной полимеразной цепной реакции.
Лечение синдрома Ди Джорджи
Специфического лечения синдрома Ди Джорджи на сегодняшний момент не существует, используют только паллиативные и симптоматические методики. Очень важно как можно раньше выявить врожденные пороки сердца и при необходимости произвести их хирургическую коррекцию, поскольку именно сердечно-сосудистые нарушения являются наиболее частой причиной неонатальной cмepти при этом заболевании. Значительную опасность представляют собой судорожные приступы, обусловленные гипокальциемией, что требует своевременной коррекции электролитного баланса плазмы крови. Помощь хирургов при синдроме Ди Джорджи также может потребоваться для устранения пороков развития лица и неба.
Из-за выраженного иммунодефицита любые признаки бактериальной, вирусной или грибковой инфекции являются поводом для срочного применения соответствующих препаратов (антибиотиков, противовирусных и фунгицидных средств). Для улучшения иммунного статуса больного синдромом Ди Джорджи может производиться заместительное вливание иммуноглобулинов, полученных из донорской плазмы. В отдельных случаях осуществлялась пересадка вилочковой железы, которая стимулировала образование собственных Т-лимфоцитов – это способствовало улучшению качества жизни больных.
Прогноз и профилактика синдрома Ди Джорджи
Прогноз синдрома Ди Джорджи большинством исследователей оценивается как неопределенный, так как данное заболевание характеризуется значительной вариабельностью симптомов. В тяжелых случаях имеется высокий риск ранней неонатальной cмepти из-за сочетания сердечно-сосудистых и иммунологических нарушений. Более доброкачественные формы синдрома Ди Джорджи требуют достаточно интенсивной паллиативной терапии, особенно важно уделять внимание лечению и профилактике вирусных и грибковых инфекций. Интеллектуальное развитие больных несколько замедлено, однако при правильной педагогической и психологической коррекции проявления задержки развития можно нивелировать. Из-за частого спонтанного характера мутаций профилактика синдрома Ди Джорджи не разработана.
Синдром Ди Джорджи
. или: Синдром Ди Георга, велокардиофациальный синдром, синдром третьего и четвертого фарингеальных мешков, синдром конотрункальной аномалии лица, 22q112 делеции синдром
Синдром Ди Джорджи — это первичный (врожденный, возникший внутриутробно или передавшийся от родителей к детям) иммунодефицит (снижение иммунитета), для которого характерны аплазия (отсутствие) или гипоплазия (уменьшение размеров) тимуса (вилочковой железы) и паращитовидных желез, врожденные пороки сердца, лицевые мальформации (изменения). Также заболевание может сопровождаться другими аномалиями (нарушениями) развития (аномалиями скелета, почек, нервной системы, патологией (болезнью) глаз).
При отсутствии или, что встречается гораздо чаще, уменьшении размеров тимуса Т-лимфоциты (клетки иммунной системы) не развиваются должным образом. Из-за этого иммунная система не может выполнять свои защитные функции в полном объеме. Однако « полный» синдром Ди Джорджи с выраженными аномалиями иммунной системы встречается крайне редко. Ввиду многообразия симптомов эти пациенты могут наблюдаться врачами разных специальностей.
Симптомы синдром ди джорджа
Клинические проявления (симптомы) варьируются у разных пациентов. Чаще всего встречаются следующие.
- Врожденные (возникшие внутриутробно или передавшиеся от родителей к детям) пороки сердца (вплоть до тетрады Фалло — стеноза (сужения) выходного отдела правого желудочка, высокого дефекта межжелудочковой перегородки (дефекта верхней или мембранозной части перегородки), декстрапозиции (смещения аорты вправо) аорты, гипертрофии (увеличения размеров) правого желудочка). Судороги (возникают из-за неправильной работы паращитовидных желез).
- Аномалии (нарушения) лицевого скелета:
- микроцефалия (уменьшение размеров костей черепа);
- гипертелоризм (широко расставленные глаза);
- маленькие, деформированные, низкорасположенные уши;
- наличие эпиканта (вертикальной складки кожи полулунной формы (напоминающей луну в новолуние), прикрывающей внутренний угол глазной щели);
- расщелины губы и нёба;
- « готическое нёбо» (высокое нёбо);
- микрогнатия (недоразвитие челюстных костей);
- страбизм (косоглазие);
- антимонголоидный разрез глаз (разрез глаз, при котором наружные углы глазных яблок опущены вниз).
- Аномалии развития гортани, глотки, трахеи, внутреннего уха, пищевода (стенозы, укорочения).
- Аномалии центральной нервной системы: атрофия (нарушение строения) коры головного мозга (поражение многих двигательных и чувствительных функций), гипоплазия (нарушение координации движений) мозжечка.
- Аномалии желудочно-кишечного тракта: атрезия aнycа (врожденное отсутствие выходного aнaльного отверстия), aнaльные фистулы (патологические (отсутствующие в норме) каналы между прямой кишкой и другими органами).
- Патология глаз: дизгенезия передней камеры (изменение угла передней камеры глаза), колобома (дефект одной из составляющих глазного яблока (радужки, хрусталика и т.д.), при котором некоторые составляющие отсутствуют), аномалия сосудов сетчатки (как следствие, нарушение питания сетчатки).
- Аномалии развития почек: гидронефроз (наличие жидкости в полости почки), атрофия (нарушение питания и, как следствие, гибель) почек, рефлюкс (заброс мочи в вышестоящие структуры почки – почечные чашечки, лоханки, почечные синусы, паренхиму почек).
- Аномалии зубов: позднее прорезывание (в детстве), гипоплазия эмали (нарушение правильного развития), кариес (разрушение зубов).
- Аномалии скелета: полидактилия (наличие более 5-ти пальцев на конечностях), отсутствие ногтей, спонтанные переломы костей (переломы, которые происходят в тех ситуациях, в которых здоровый человек не получил бы перелом (например, оступился на подножке автобуса, поскользнулся на улице и упал), а у человека с синдромом Ди Джорджи он возникает).
- Отставание в интеллектуальном развитии.
- Отставание в моторном развитии (освоении целенаправленных двигательных актов).
- Частое возникновение вирусных, грибковых и бактериальных инфекций, плохо поддающихся стандартной терапии (терапии, обычно применяемой при этих заболеваниях) — лечению антибиотиками (противомикробными средствами), противовирусными препаратами, антимикотиками (противогрибковыми средствами).
Читать еще: Применение вазопрессина при несахарном диабете
Причиной возникновения синдрома Ди Джорджи является делеция (выпадение участка) 22-ой хромосомы.
Возможными факторами риска появления делеции являются сахарный диабет (нарушение обмена глюкозы (сахара) в организме) у матери, употрeбление алкоголя во время беременности, вирусные заболевания в первом триместре беременности.
Есть данные, что поврежденная 22-ая хромосома может наследоваться по аутосомно-доминантному типу, то есть заболевание передается человеку от одного из родителей.
Врач терапевт поможет при лечении заболевания
Диагностика
- Анализ анамнеза заболевания и жалоб – выраженный кариес (поражение зубов); переломы; проблемы с сердцем; часто рецидивирующие (повторяющиеся) бактериальные, вирусные и грибковые заболевания, трудно поддающиеся лечению.
- Анализ анамнеза жизни —отставание в росте и развитии ребенка; наличие пороков сердца, были ли операции по этому поводу, наличие косоглазия, были ли операции по этому поводу; частые бактериальные, вирусные и грибковые инфекции.
- Осмотр пациента – врач может увидеть следующую картину: уменьшение головы пациента, гипертелоризм (широко расставленные глаза), маленькие, деформированные, низкорасположенные уши, наличие эпиканта (вертикальной складки кожи полулунной формы, прикрывающей внутренний угол глазной щели), расщелины губы и нёба, « готическое нёбо» (высокое нёбо), микрогнатию (недоразвитие челюстных костей), страбизм (косоглазие), антимонголоидный разрез глаз (разрез глаз, при котором наружные углы глазных яблок опущены вниз). При аускультации (прослушивании) сердца можно услышать специфические шумы, характерные для врожденных (возникших внутриутробно из-за хромосомных нарушений, характерных для этого заболевания) пороков сердечно-сосудистой системы.
- Иммунный статус –для данного анализа берется кровь из вены; определяется снижение количества Т- лимфоцитов (клеток иммунной системы), может выявляться снижение уровня сывороточных иммуноглобулинов (белков, защищающих организм от чужеродных агентов (бактерий, вирусов, грибов)).
- Общий анализ крови — лимфопения (уменьшение количества лимфоцитов).
- Биохимический анализ крови —снижение уровня кальция, гипокальциемия (анализ повторяют несколько раз, чтобы убедиться в стойком наличии этого состояния).
- Ультразвуковое исследование (УЗИ) паращитовидных желез и тимуса (вилочковой железы) — выявляется их отсутствие или уменьшение.
- Эхокардиография(ЭхоКГ)сердца— выявление пороков сердечно-сосудистой системы.
- Флюоресцентная гибридизация ДНК —позволяет выявить делеции (участки выпадения) в 22-ой хромосоме, характерные для синдрома Ди Джорджи.
- Возможна также консультация аллерголога-иммунолога.
Лечение синдром ди джорджа
- Антибиотики (противомикробные средства) назначаются при возникновении бактериального инфекционного процесса в организме.
- Противовирусные препараты — при возникновении вирусной инфекции.
- Противогрибковые препараты (антимикотики) — при грибковой инфекции.
- Заместительная (назначаемая при недостатке своих) терапия внутривенными иммуноглобулинами, полученными из плазмы крови (жидкой части крови) здоровых доноров, которая применяется в случае снижения уровня иммуноглобулинов (белков, защищающих организм от чужеродных агентов (бактерий, вирусов, грибов)).
- Препараты кальция с целью увеличения его уровня.
- Хирургическое лечение — устранение врожденных (возникших внутриутробно или передавшихся от родителей) пороков сердечно-сосудистой системы.
- Трансплантация (пересадка) фетального тимуса (вилочковой железы) без предварительной хирургической коррекции врожденных пороков сердца считается неэффективной, проводится только при « полном» синдроме ДиДжорджи (когда есть выраженные иммунологические нарушения – тяжелый иммунодефицит (сильное снижение иммунитета)).
Осложнения и последствия
- Значительное отставание в интеллектуальном развитии.
- Развитие аутоиммунных заболеваний (для этих болезней характерна агрессия иммунной системы против своего же организма: иммунная система принимает свои клетки за чужеродные и атакует их).
- Развитие опухолевых заболеваний в раннем возрасте.
- Летальный исход (cмepть) от инфекционных осложнений или пороков сердечно-сосудистой системы, не совместимых с жизнью, эндокринной патологии (нарушения функции паращитовидных желез).
Прогноз обычно зависит от выраженности кардиологических и эндокринологических дефектов, при « полном» синдроме – от иммунологических (насколько выражен иммунодефицит (ослабление иммунитета) – отсутствие Т-лимфоцитов (клеток иммунной системы), снижение продукции (выработки) антител – иммуноглобуллинов (белков, защищающих организм от чужеродных агентов (бактерий, вирусов, грибов)).
Профилактика синдром ди джорджа
- Развитие опухолевых заболеваний в раннем возрасте.
- Пациентам с частичными иммунными нарушениями может назначаться профилактическая антибактериальная (противомикробная) и противогрибковая терапия.
- Необходимо исключить употрeбление алкоголя во время беременности матери.
- До начала беременности матери необходимо сделать соответствующие противовирусные прививки (например, от вируса краснухи (вирусного заболевания, не представляющего опасности для взрослого человека, но способного вызвать серьезные нарушения в нервной системе плода) и кори (тяжелого вирусного заболевания, представляющего опасность для жизни и здоровья беременной женщины и плода)).
Если беременной женщине во время планового скрининга (проведения ультразвукового исследования плода и органов малого таза на 11-13 недели беременности) на наличие патологии (заболевания) предположили возможное наличие у ребенка синдрома Ди Джорджи, то ей предлагают пройти дополнительное обследование — амниоцентез (получение околоплодных вод) с целью проведения анализа ДНК плода на предмет хромосомных нарушений (делеция (выпадение участка) 22-ой хромосомы).
- Кондратенко И.В., Бологов А.А. Первичные иммунодефициты – М.: ИД МЕДПРАКТИКА-М,2005 – 232с.
- Аллергология и иммунология. Национальное руководство. Под редакцией Р.М.Хаитова, « Гэотар-Медиа», 2009.
- Bassett A.S., Chow E.W.C., Husted J. et al. Clinical features of 78 adults with 22q11 deletion syndrome// Am.J.Med.Genet. — 2005 – N 138A – P. 307 – 313.
Что делать при синдром ди джордже?
- Выбрать подходящего врача терапевт
- Сдать анализы
- Получить от врача схему лечения
- Выполнить все рекомендации
Синдром Ди Джорджи
Синдром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) связан с дефектом развития четвертого глоточного кармана и четвертой жаберной дуги. Данный синдром имеет генетически неоднородный характер с частичной моносомией проксимального плеча 22-й хромосомы (при микроскопическом исследовании выявляется в трети случаев) и высокой частотой делеций (88%), выявленных по результатам FISH-исследований (Driscoll et al., 1993). В редких случаях заболевание сочеталось делецией 17р и 10p (Levy-Mozziconacci et al., 1994).
Уровень распространения синдрома делеции 22q11 (22Q11DS оценивается приблизительно как 3 случая на 10000 новорожденных (Oskarsdottir 2005) и возможно несколько чаще встречается среди девочек (личные неопубликованные данные).
В настоящее время большая часть практикующих врачей рассматривает синдром делеции 22q11 как общепринятый синдром с фенотипом Ди Джорджи. Таким образом, диагноз устанавливается в случае выявления «САТСН-22 фенотипа» (аномалии сердца (Cardiac), аномалии (Anomalous) лица, гипоплазии тимуса (Thymus), расщелины (Cleft) неба (включая только подслизистое расщепление) и гипокальциемии (Hypocalcaemia) и делеции сегмента 22q11, выявленной методом FISH-диагностики.
Анализ методом FISH обеих метафазных хромосом и интерфазной клетки пациента с синдромом Ди Джорджи,
демонстрирующий делецию зонда TUPLE1 (официальное название — HIRA), который локализуется на хромосоме 22q11.2.
Красным светится зонд TUPLE1, зеленым — контрольный зонд, локализующийся на 22q. Метафазная пластинка содержит одну 22-ю хромосому с зеленым и красным сигналами.
Другая 22-я хромосома содержит только зеленый сигнал, красный сигнал отсутствует в связи с делецией на этой хромосоме.
На интерфазной клетке также определяются две области гибридизации: зеленая и красная, что иллюстрирует делецию на хромосоме 22q11.2.
а) Клинические проявления. Тимус и паратиреоидные железы отсутствуют или эктопированы, также часто отмечается мальформация крупных сосудов основания сердца. Отсутствие тимуса может сопровождаться тяжелым иммунодефицитом, а отсутствие паращитовидных желез—тяжелыми гипокальциемическими судорогами в период новорожденности и позже. Гипокальциемия связана с паратиреоидным гормоном. Часто встречается расщелина неба, микрогнатия, низко расположенные уши и гипертелоризм (Conley et al., 1979).
Частично совпадающий фенотип, известный как вело-кардио-фациальный синдром или синдром Шпринтцена, проявляется сходными аномалиями сердца, дисморфизмом лица, расщелиной неба и задержкой умственного развития, иногда сопровождается поздним развитием психиатрических отклонений и также сочетается с делецией 22q11. Также может отмечаться гипоплазия мозжечка (Devriendt et al., 1996).
Синдром делеции сегмента 22q11 проявляется достаточно типичным поведенческим фенотипом. Отмечается задержка развития разговорной речи, но некоторые вербальные навыки, в частности словарный запас, имеют тенденцию к лучшему развитию, чем функции, связанные с достижением школьного возраста. Коэффициент IQ обычно составляет около 70, у отдельных пациентов превышает 100 и (очень редко) не достигает 40. Неуклюжесть движений является типичным проявлением данного заболевания, и клинические проявления синдрома делеции сегмента 22q11 часто полностью соответствуют критериям нарушения развития координации.
Синдром Ди Джорджи
Характерен упадок сил, который в большинстве случаев отмечается с самого раннего возраста, в связи с чем для начала любых даже самых простых действий пациента необходимо «заводить», что противоречит относительно «хорошему» (по сравнению с другими синдромами с поведенческим фенотипом) IQ. Большая часть пациентов «не успевает» в школе; это касается даже немногочисленной группы с IQ около 100 (Oskarsdottir et al., 2005). У многих пациентов выявляются критерии синдрома гиперактивности с дефицитом внимания (СГДВ), в частности, смешанного подтипа и подтипа с дефицитом внимания. С возрастом у большей части пациентов подтип СДГВ сменяется на подтип с дефицитом внимания (Niklasson et al., 2005). Типичны аутистические проявления (Campbell et al„ 2006), но истинный аутизм встречается редко.
Даже в случаях, когда признаки аутизма отсутствуют, у пациентов отмечаются социальные проблемы и проблемы общения. Данные отклонения часто сопровождаются растягиванием слов, проблемами артикуляции и бедной мимикой, характерной для синдрома делеции сегмента 22q11, так как во многих случаях отмечается небно-глоточная недостаточность. Депрессивное настроение часто отмечается, начиная с подросткового возраста. Психозы могут развиваться в позднем подростковом возрасте и раннем взрослом возрасте, даже в случаях с минимальными психиатрическими отклонениями в детском возрасте (Sobin et al., 2005). СДГВ и расстройства группы аутизма при синдроме делеции сегмента 22q11 могут быть результатом патологии головного мозга с уменьшением серого вещества коры, большого хвостатого ядра и аномалии белого вещества лобных долей и мозжечка. В подростковом и взрослом возрасте увеличивается риск развития шизофрении (Niklasson et al., 2002).
Читать еще: Белые гнойные пробки в миндалинах без температуры и с ней — что это?
б) Лечение синдрома Ди Джорджи. Как и в случае других синдромов с поведенческим фенотипом важен ранний диагноз и обучение семьи психологической помощи. СДГВ в некоторых случаях успешно поддается лечению стимуляторами и методами когнитивно-поведенческой терапии в сочетании со специальными программами образования. Ингибиторы захвата серотонина или комбинированного захвата серотонина и норадреналина можно попробовать при длительных периодах депрессивного настроения или дистимии. Психоз (в случае его развития) может с трудом подаваться лечению, а типичные и атипичные нейролептики, несмотря на их эффективность в отношении психотических симптомов, а иногда и слуховые галлюцинации, часто менее эффективны, чем в случае других психозов у молодых людей.
Редактор: Искандер Милевски. Дата публикации: 4.12.2018
Проявления и лечение синдрома Ди Джорджи
Синдром Ди Джорджи – генетическое заболевание, возникающее на фоне повреждения хромосомного набора человека. Патология выявляется в первые месяцы жизни ребенка. Симптомы проблемы включают в себя изменение нормального строения черепа, врожденные пороки сердца, частые и длительные инфекционные поражения на фоне первичного иммунодефицита. Нарушение работы защитных сил организма возникает вследствие недоразвитости или полного отсутствия тимуса, а также деформации паращитовидных желез. Диагностика синдрома Ди Георга основана как на клинической картине, так и на результатах лабораторных тестов. Лечение предполагает использование медикаментозных средств, а также хирургическое вмешательство для коррекции сформировавшихся нарушений.
Причины возникновения синдрома
Заболевание является патологией, обусловленной генетически. Оно формируется на фоне повреждения участка 22-ой хромосомы. Данный фрагмент кодирует несколько важных факторов, обеспечивающих процесс деления клеток в ходе эмбриогенеза, то есть развития плода в организме матери. Изменения в этой структуре приводят к нарушению закладки фарингеальных мешков, из которых в дальнейшем формируется нёбо, тимус, а также паращитовидные железы. Кроме дефекта в структуре 22-ой хромосомы, сходные клинические признаки синдрома Ди Джорджи возникают и на фоне других генетических изменений.
Причины, по которым формируются дефекты в строении ДНК, неясны. Предполагается роль провоцирующих факторов: вредных привычек, радиации, а также употрeбления алкоголя, сильнодействующих лекарственных средств и курения во время беременности.
Основные признаки патологии
Клиническая картина заболевания связана с пороками развития эндокринных органов и лица. При этом часть проявлений можно выявить еще в ходе пренатальной диагностики, другие же определяются только после рождения ребенка. Симптомы синдрома Ди Джорджи включают в себя:
- Расщепление мягкого и твердого неба. Подобный дефект зачастую сочетается с другой аномалией – «заячьей губой». Интенсивность выраженности данной проблемы значительно варьируется. В ряде случаев расщелина мешает детям нормально питаться, а у некоторых пациентов диагностируется только при тщательном осмотре и не доставляет малышам неудобств.
- Зубы и прикус также страдают при синдроме Ди Джорджи. Распространенным симптомом является прогнатия, когда верхняя челюсть становится чрезмерно выдвинута вперед по сравнению с нижней. Это затрудняет прием пищи, а также повышает риск формирования стоматологических заболеваний. У пациентов замедляется прорезывание зубов, быстро развивается кариес.
- Первичный иммунодефицит, являющийся основным проявлением болезни, формируется вследствие недоразвитости или полного отсутствия тимуса, сопровождается возникновением тяжелых симптомов. Дети часто болеют, при этом многие инфекционные и вирусные патологии способны вызвать формирование осложнений. Распространены среди пациентов и грибковые поражения, позволяющие заподозрить расстройство функции защитных сил организма.
- Недостаточное развитие паращитовидных желез приводит к дефициту кальция в крови, что сопровождается возникновением судорог. У некоторых пациентов формируется тетания мышц. Данный симптом проявляется уже через несколько дней после рождения ребенка. Отличительной чертой этого признака является отсутствие ответа на введение противосудорожных препаратов. Без лечения данное состояние приводит к гибели пациента.
- Многие дети с синдромом Ди Георга страдают от врожденных пороков сердца и крупных сосудов, что сопровождается угрожающими жизни симптомами и значительно ухудшает прогноз заболевания. У малышей быстро развивается гипоксия, проявляющаяся одышкой, цианозом кожи и слизистых, тахикардией. Самыми распространенными нарушениями являются дефекты строения аорты, а также развитие признаков тетрады Фалло. Возникновение симптомов сердечной недостаточности требует оказания неотложной помощи, а также проведения хирургического лечения.
- У некоторых детей синдром сопровождается умственной отсталостью, возникающей вследствие атрофии коры головного мозга. Недоразвитость мозжечка приводит к нарушению координации, парезам и даже параличам. В старшем возрасте пациенты страдают от эмоциональной нестабильности, речевых дисфункций и психозов различной интенсивности.
Принято считать, что синдром Ди Георга относится к числу редких заболеваний. Однако существуют данные, указывающие на то, что эта проблема является одной из самых распространенных генетических патологий, сопровождающихся делецией хромосом. Неадекватность информации о частоте встречаемости расстройства ученые связывают с двумя факторами: разнообразием фенотипов поражения, то есть вариаций клинических признаков, а также недостаточно тщательной диагностикой.
Врачи, занимающиеся статистической обработкой распространенности синдрома Ди Джорджи, призывают к настороженности в отношении заболевания. Рекомендуется исследовать пациентов с врожденными пороками сердца на наличие генетических аномалий, особенно в тех случаях, когда формируются послеоперационные осложнения. Требуется также проведение гематологических тестов у младенцев.
Общий клинический анализ крови, относящийся к числу доступных методов диагностики, позволяет вывить лимфопению. Этот признак может быть использован при раннем выявлении синдрома Ди Георга, что позволяет предотвратить стремительное развитие симптомов поражения. Биохимический тест используется с целью оценки уровня кальция в крови новорожденных, так как возникновение судорог на фоне снижения концентрации этого соединения относится к числу опасных проявлений генетической аномалии.
Используемые методы диагностики
После рождения ребенка происходит развитие специфических симптомов, позволяющих заподозрить синдром Ди Джорджи. Присутствие в анамнезе постоянно рецидивирующих длительно протекающих инфекций в сочетании с деформациями лица или пороками сердца указывает на наличие заболевания. Диагностика подразумевает проведение анализов крови. Отмечается снижение факторов клеточного иммунитета, лимфопения, а также уменьшение концентрации кальция с одновременным повышением уровня фосфора. УЗИ используется как для выявления дефектов в строении сердца и крупных сосудов, так и для оценки структуры тимуса и паращитовидных желез.
С помощью МРТ удается получить более точные фото внутренних органов, которые используются при дифференциации патологии от расстройств со сходной симптоматикой. Точный диагноз ставится на основании проведения генетического анализа. При этом могут обнаруживаться несколько вариантов мутаций, самой распространенной из которых является изменение структуры 22-ой хромосомы.
В ряде клинических примеров у пациентов с синдромом Ди Джорджи удалось выявить проблему еще на этапе пренатального скрининга. Как правило, проведение УЗИ на ранних сроках беременности неинформативно, однако ближе к концу удается выявить пороки развития клапанного аппарата сердца, а также характерные деформации костей черепа. Самым точным методом диагностики заболевания до рождения является амниоцентез. Эта процедypa представляет собой забор околоплодных вод для анализа генетического материала ребенка.
Проблема заключается в травматичности данной техники и высоком риске развития опасных осложнений. При этом точность анализа достигает 99% процентов. Следовательно, если синдром Ди Джорджи не подтвердился при амниоцентезе, проблема исключается из списка дифференциальных диагнозов.
Необходимое лечение
Борьба с недугом носит симптоматический характер. Она направлена на коррекцию сформировавшихся нарушений и предупреждение развития осложнений. Используются как консервативные, так и хирургические техники.
Медикаментозная терапия синдрома Ди Георга предполагает назначение следующих препаратов:
- Антибактериальные средства применяются с целью борьбы с инфекционными поражениями. Часто используются пенициллины, например, «Амоксициллин», а также цефалоспорины, к которым относится «Цефазолин».
- Если ребенок страдает от вирусных заболеваний, назначаются такие медикаменты, как «Ацикловир» и «Цитовир».
- Важную роль играет применение препаратов кальция. Только данная терапия позволяет бороться с судорогами и предотвратить развитие фатальных осложнений. Применяются как инъекционные формы, так и добавки к пище, что зависит от степени выраженности недостатка элемента в крови. Используются такие средства, как «Кальцемин».
- Для коррекции нарушений со стороны нервной системы применяются седативные медикаменты. Начинают с препаратов растительного происхождения, например, «Персена», а в случае отсутствия эффекта прибегают к использованию транквилизаторов, таких как «Диазепам».
Хирургия направлена в первую очередь на коррекцию работы сердечно-сосудистой системы. Проводится оперативное лечение врожденных пороков кардиальных структур. Только после стабилизации состояния пациента задумываются о трaнcплантации тимуса или паращитовидных желез. В ряде случаев применяются также хирургические техники, направленные на коррекцию дефектов лицевых костей черепа.
Возможные осложнения и прогноз
Самыми опасными для пациентов с синдромом Ди Джорджи являются последствия сердечной недостаточности. Они способны приводить к отеку легких и гибели ребенка. Судороги также относятся к числу осложнений при заболевании. Инфекции у малышей с недугом протекают тяжело. Возможно развитие негативных последствий со стороны дыхательной системы, пищеварительного тракта и слухового анализатора.
Исход заболевания определяется выраженностью патологических изменений. Прогноз при поражении от осторожного до нeблагоприятного.
Рекомендации по профилактике
Специфических мер, позволяющих предотвратить возникновение проблемы, не разработано. Будущим мамам следует уделять внимание правильному планированию и ведению беременности. Необходимо правильно питаться, отказаться от вредных привычек. Профилактика недуга подразумевает также поддержание естественной работы иммунитета за счет недопущения переохлаждения, ограничения контактов с потенциальными источниками инфекции и своевременной вакцинации.
Отзывы о лечении
Петр, 36 лет, г. Москва
Дочь родилась с редким генетическим заболеванием – синдромом Ди Джорджи. Врачи сказали, что у нее легкая форма, так что нам повезло. Правда, все равно пришлось сделать операцию по исправлению расщелины в небе. После хирургического вмешательства дочка долго принимала антибиотики, чтобы не подхватить инфекцию.
Жанна, 29 лет, г. Екатеринбург
У ребенка при рождении диагностировали синдром Ди Джорджи, из-за которого развился порок сердца, были изъяны в чертах лица и иммунодефицит. Его прооперировали, долго держали в стационаре. После выписки сын подхватил серьезную инфекцию, которая поразила легкие. Несмотря на лечение, помочь малышу не удалось.
Важно знать родителям о здоровье:
FitoSpray для похудения (Фитоспрей)
")
FitoSpray для похудения ( Фитоспрей) FitoSpray — спрей для похудения Многие мечтают похудеть, стать стройными, обрести фигуру мечты. Неправильное питание,…
30 04 2023 21:23:46
Фитостеролы в продуктах питания

Фитостеролы в продуктах питания Фитостерины Существует много питательных веществ, которые, как утверждают исследователи, могут положительно повлиять на…
29 04 2023 11:47:13
Фитотерапевт

Фитотерапевт Фитотерапевт Я, Ирина Гудаева — травница, массажист, ведущая семинаров по созданию натуральной косметики и курса « Практическое травоведение»…
28 04 2023 11:53:40
Fitvid

Fitvid Брекеты: минусы, трудности, проблемы Брекет-системы помогли избавиться от комплексов миллионам людей. Это действительно эффективный инструмент,…
27 04 2023 3:11:21
Фониатр

Фониатр Фониатрия – один из разделов медицины. Фониатры изучают патологии голоса, методы их лечения, профилактики, а также способы коррекции…
24 04 2023 6:39:23
Форель

Форель Форель относится к отряду лососеобразных, семейству лососевых. Ее тело удлинено, немного сжато с боков, покрыто мелкой чешуей. Замечательной…
23 04 2023 18:23:20
Формула идеального веса

Формула идеального веса Калькулятор нормы веса Вес 65 кг относится к категории Норма для взрослого человека с ростом 170 см . Эта оценка основана на…
20 04 2023 20:57:28
Формулы расчета идеального веса

Формулы расчета идеального веса Фoрмулa «идeальнoго вeса» То, что ожирение шагает семимильными шагами по планете – это факт. И, несмотря на то, что…
19 04 2023 21:47:15
Фосфатида аммонийные соли

Фосфатида аммонийные соли Аммонийные соли фосфатидиловой кислоты ( Е442) Е442 – это пищевая добавка, которую относят к категории эмульгаторов. Вещество…
18 04 2023 8:32:11
Фототерапия новорожденных

Фототерапия новорожденных Фототерапия новорожденных Применение фототерапии для новорожденных С момента своего рождения организм ребенка начинает адаптацию…
17 04 2023 0:33:51
Фототерапия новорожденных при желтухе

Фототерапия новорожденных при желтухе Фототерапия новорожденных После появления ребенка на свет его организм адаптируется к совершенно иным условиям…
16 04 2023 5:48:36
Французская диета

Французская диета Французская диета Эффективность: до 8 кг за 14 дней Сроки: 2 недели Стоимость продуктов: 4000 рублей на 14 дней Общие правила…
15 04 2023 21:47:57
Фрукт Кумкват — что это такое?

Фрукт Кумкват — что это такое? Фрукт Кумкват — что это такое? Впервые упоминают необычный для европейцев фрукт китайские летописи 11 века. Португальские…
14 04 2023 12:49:49
Фруктовая диета

Фруктовая диета Фруктовая диета Эффективность: 2-5 кг за 7 дней Сроки: 3-7 дней Стоимость продуктов: 840-1080 рублей в неделю Общие правила Фруктовая…
12 04 2023 16:16:52
Фруктоза при диабете

Фруктоза при диабете Можно ли фруктозу при сахарном диабете? Для многих диабет является той проблемой, которая вносит в жизнь ряд ограничений. Так, к…
09 04 2023 9:42:38
Фрукт свити – польза и вред

Фрукт свити – польза и вред Свити — что это за фрукт? Что такое свити? Продолжаем разбирать цитрусовые, но как всегда идем не по верхам, а копаем глубже и…
06 04 2023 23:45:47
Фрукты и ягоды

Фрукты и ягоды Разница между фруктом и ягодой Фрукты и ягоды любят практически все. Ведь они такие вкусные и полезные! Мы любуемся лежащими на столе…
05 04 2023 8:58:54
Фтизиатр

Фтизиатр Врачи фтизиатры Москвы Фтизиатр — это дипломированный специалист в области фтизиатрии. Он специализируется на профилактике, диагностике, лечении…
03 04 2023 9:50:35
Фтор в организме человека

Фтор в организме человека Фтор в организме человека Дневная норма потрeбления Мужчины старше 60 лет Женщины старше 60 лет Беременные (2-я половина)…
02 04 2023 12:52:14
Боли в спине после рождения ребёнка

Боли в спине после рождения ребёнка Почему после родов болит спина У мамочек нередко болит спина после родов. Причем, дискомфорт может длиться довольно…
30 03 2023 18:38:59
Фунчоза: польза и вред

Фунчоза: польза и вред Фунчоза: польза и возможный вред Увлечение восточной кухней год от года растет. Принято считать, что такой рацион полезен для…
29 03 2023 15:54:45
Фундук

Фундук В рационе здорового человека обязательно присутствуют орехи в различных вариациях. Среди них выгодно выделяется фундук. Высокая пищевая ценность и…
28 03 2023 3:25:17
Фуросемид таблетки инструкция по применению

Фуросемид таблетки инструкция по применению Инструкция по применению: Цены в интернет-аптеках: Фуросемид – синтетическое диуретическое лекарственное…
25 03 2023 18:12:40
Галактоза

Галактоза Галактоза – это представитель класса простых молочных сахаров. В человеческий организм поступает преимущественно в составе молока,…
24 03 2023 1:53:48
Галанга

Галанга С древних времен растения играют важную роль в жизни человека, в том числе и для поддержания здоровья. Некоторые травы известны как лучшие…
23 03 2023 14:13:12
Галега лекарственная

Галега лекарственная Галега лекарственная (Galega officinalis) Син: козлятник лекарственный, козлятник аптечный, козья рута, французская сирень, солодянка…
22 03 2023 2:37:27
Боли в суставах при беременности

Боли в суставах при беременности Боли в суставах при беременности В период беременности у женщины могут возникать различные боли в самых разных местах….
19 03 2023 10:28:24
Гастрит и изжога

Гастрит и изжога Лучшие лекарства от изжоги и гастрита Многие пациенты с гастритом и другими заболеваниями Ж К Т страдают от изжоги. Данный симптом может…
18 03 2023 1:15:15
Где находится ключица у человека на фото?

Где находится ключица у человека на фото? Ключица человека: анатомия, строение, функции Ключица – это единственное костное образование в теле человека,…
15 03 2023 22:27:10