Серповидно-клеточная анемия (S-гемоглобинопатия)

Серповидно-клеточная анемия – наследственная гемоглобинопатия, обусловленная синтезом аномального гемоглобина S, изменением формы и свойств эритроцитов крови. Серповидно-клеточная анемия проявляется гемолитическими, апластическими, секвестрационными кризами, тромбозами сосудов, костно-суставными болями и припухлостью конечностей, изменениями скелета, сплено- и гепатомегалией. Диагноз подтверждается по данным исследования периферической крови и пунктата костного мозга. Лечение серповидно-клеточной анемии является симптоматическим, направленным на предупреждение и купирование кризов; может быть показано переливание эритроцитов, прием антикоагулянтов, проведение спленэктомии.
Общие сведения
Серповидно-клеточная анемия (S-гемоглобинопатия) – разновидность наследственной гемолитической анемии, характеризующаяся нарушением структуры гемоглобина и присутствием в крови эритроцитов серповидной формы. Заболеваемость серповидно-клеточной анемией распространена, главным образом, в странах Африки, Ближнего и Среднего Востока, Средиземноморского бассейна, Индии. Здесь частота носительства гемоглобина S среди коренного населения может достигать 40%. Любопытно, что больные серповидно-клеточной анемией имеют повышенную врожденную устойчивость к заражению малярией, поскольку малярийный плазмодий не может проникнуть в эритроциты серповидной формы.

Серповидно-клеточная анемия
Причины
В основе серповидно-клеточной анемии лежит генная мутация, обусловливающая синтез аномального гемоглобина S (HbS). Дефект структуры гемоглобина характеризуется заменой глутаминовой кислоты валином в ß-полипептидной цепи. Образующийся при этом гемоглобин S после потери присоединенного кислорода приобретает консистенцию высокополимерного геля и становится в 100 раз менее растворимым, чем нормальный гемоглобин А. В результате этого эритроциты, несущие деоксигемоглобин S, деформируются и приобретают характерную полулунную (серповидную) форму. Измененные эритроциты становятся ригидными, малопластичными, могут закупоривать капилляры, вызывая ишемию тканей, легко подвергаются аутогемолизу.
Наследование серповидно-клеточной анемии происходит по аутосомно-рецессивному типу. При этом, гетерозиготы наследуют дефектный ген серповидно-клеточной анемии от одного из родителей, поэтому, наряду с измененными эритроцитами и HbS, имеют в крови и нормальные эритроциты с HbА. У гетерозиготных носителей гена серповидно-клеточной анемии признаки заболевания возникают лишь в определенных условиях. Гомозиготы наследуют по одному дефектному гену от матери и от отца, поэтому в их крови присутствуют только серповидные эритроциты с гемоглобином S; заболевание развивается рано и протекает тяжело.
Таким образом, в зависимости от генотипа, в гематологии различают гетерозиготную (HbAS) и гомозиготную (HbSS, дрепаноцитоз) форму серповидно-клеточной анемии. К редко встречающимся вариантам заболевания относятся промежуточные формы серповидно-клеточной анемии. Обычно они развиваются у двойных гетерозигот, несущих один ген серповидно-клеточной анемии и другой дефектный ген — гемоглобина C (HbSC), серповидной β-плюс (HbS/β +) или β-0 (HbS/β0) талассемии.
Симптомы серповидно-клеточной анемии
Гомозиготная серповидно-клеточная анемия обычно проявляется у детей к 4-5 месяцу жизни, когда увеличивается количество HbS, а процентное содержание серповидных эритроцитов достигает 90%. В таких случаях раннее возникновение гемолитической анемии у ребенка обуславливает задержку физического и умственного развития. Характерны нарушения развития скелета: башенный череп, утолщение лобных швов черепа в виде гребня, кифоз грудного или лордоз поясничного отдела позвоночника.
В развитии серповидно-клеточной анемии выделяют три периода: I — с 6 месяцев до 2-3 лет, II — с 3 до 10 лет, III — старше 10 лет. Ранними сигналами серповидно-клеточной анемии служат артралгии, симметричное опухание суставов конечностей, боли в груди, животе и спине, желтушность кожи, спленомегалия. Дети с серповидно-клеточной анемией относятся к категории часто болеющих. Степень тяжести течения серповидно-клеточной анемии тесно коррелирует с концентрацией HbS в эритроцитах: чем она выше, тем тяжелее выражена симптоматика.
В условиях интеркуррентной инфекции, стрессовых факторов, обезвоживания, гипоксии, беременности и пр. у больных данным видом наследственной анемии могут развиваться серповидно-клеточные кризы: гемолитический, апластический, сосудисто-окклюзионный, секвестрационный и др.
При развитии гемолитического криза состояние больного резко ухудшается: возникает фебрильная лихорадка, в крови повышается непрямой билирубин, усиливается желтушность и бледность кожных покровов, появляется гематурия. Стремительный распад эритроцитов может привести к анемической коме. Апластические кризы при серповидно-клеточной анемии характеризуются угнетением эритроидного ростка костного мозга, ретикулоцитопенией, снижением гемоглобина.
Следствием депонированием крови в селезенке и печени служат секвестрационные кризы. Они сопровождаются гепато- и спленомегалией, сильными болями в животе, резкой артериальной гипотонией. Сосудисто-окклюзионные кризы протекают с развитием тромбоза сосудов почек, ишемии миокарда, инфаркта селезенки и легких, ишемического приапизма, окклюзии вен сетчатки, тромбоза мезентериальных сосудов и др.
Гетерозиготные носители гена серповидно-клеточной анемии в обычных условиях ощущают себя практически здоровыми. Морфологически измененные эритроциты и анемия у них возникают только в ситуациях, связанных с гипоксией (при тяжелой физической нагрузке, авиаперелетах, восхождении в горы и др.). Вместе с тем, остро развившийся гемолитический криз при гетерозиготной форме серповидно-клеточной анемии может иметь летальный исход.

Симметричное опухание суставов при серповидно-клеточной анемии
Осложнения
Хроническое течение серповидно-клеточной анемии с повторными кризами приводит к развитию целого ряда необратимых изменений, нередко становящихся причиной гибели больных. Примерно у трети больных отмечается аутоспленэктомия – сморщивание и уменьшение размеров селезенки, вызванное замещением функциональной ткани рубцовой. Это сопровождается изменением иммунного статуса больных серповидно-клеточной анемией, более частым возникновением инфекций (пневмонии, менингита, сепсиса и др.).
Исходом сосудисто-окклюзионных кризов могут стать ишемические инсульты у детей, субарахноидальные кровоизлияния у взрослых, легочная гипертензия, ретинопатия, импотенция, почечная недостаточность. У женщин с серповидно-клеточной анемией отмечается позднее становление менструального цикла, склонность к самопроизвольному прерыванию беременности и преждевременным родам. Следствием ишемии миокарда и гемосидероза сердца служит возникновение хронической сердечной недостаточности; повреждения почек — хронической почечной недостаточности.
Длительный гемолиз, сопровождаемый избыточным образованием билирубина, приводит к развитию холецистита и желчнокаменной болезни. У больных серповидно-клеточной анемией часто возникают асептические некрозы костей, остеомиелит, язвы голеней.
Диагностика
Диагноз серповидно-клеточной анемии выставляется гематологом на основании характерных клинических симптомов, гематологических изменений, семейно-генетического исследования. Факт наследования ребенком серповидно-клеточной анемии может быть подтвержден еще на этапе беременности с помощью биопсии ворсин хориона или амниоцентеза.
В периферической крови отмечается нормохромная анемия (1-2х1012/л), снижение гемоглобина (50-80 г/л), ретикулоцитоз (до 30%). В мазке крови обнаруживаются серповидно измененные эритроциты, клетки с тельцами Жолли и кольцами Кабо. Электрофорез гемоглобина позволяет определить форму серповидно-клеточной анемии – гомо- или гетерозиготную. Изменение биохимических проб крови включает гипербилирубинемию, увеличение содержания сывороточного железа. При исследовании пунктата костного мозга выявляется расширение эритробластического ростка кроветворения.
Дифференциальная диагностика направлена на исключение других гемолитических анемий, вирусного гепатита А, рахита, ревматоидного артрита, туберкулеза костей и суставов, остеомиелита и др.

Серповидно-клеточная анемия
Лечение серповидно–клеточной анемии
Серповидно-клеточная анемия относится к категории неизлечимых болезней крови. Таким пациентам требуется пожизненное наблюдение гематолога, проведение мероприятий, направленных на предупреждение кризов, а при их развитии – проведение симптоматической терапии.
В период развития серповидно-клеточного криза требуется госпитализация. С целью быстрого купирования острого состояния назначается кислородотерапия, инфузионная дегидратация, введение антибиотиков, обезболивающих средств, антикоагулянтов и дезагрегантов, фолиевой кислоты. При тяжелом течении обострений показано переливание эритроцитарной массы. Проведение спленэктомии не способно повлиять на течение серповидно-клеточной анемии, однако может на время уменьшить проявления заболевания.
Прогноз и профилактика
Прогноз гомозиготной формы серповидно-клеточной анемии неблагоприятный; большая часть пациентов погибает в первое десятилетие жизни от инфекционных или тромбоокклюзионных осложнений. Течение гетерозиготных форм патологии гораздо более обнадеживающее.
Для предупреждения быстро прогрессирующего течения серповидно-клеточной анемии следует избегать провоцирующих условий (обезвоживания, инфекций, перенапряжения и стрессов, экстремальных температур, гипоксии и пр.). Детям, страдающим данной формой гемолитической анемии, в обязательном порядке показана вакцинация против пневмококковой и менингококковой инфекции. При наличии в семье больных серповидно-клеточной анемией необходима медико-генетическая консультация для оценки риска развития заболевания у потомства.
Серповидно-клеточная анемия — лечение в Москве
| Sickle cell disease | |
|---|---|
| Other names | Sickle cell disorder |
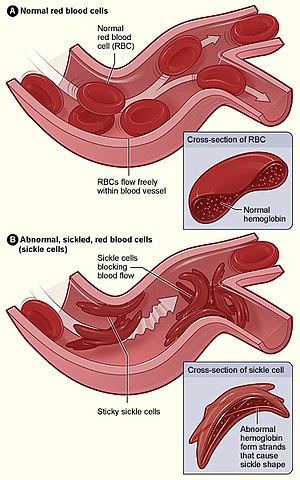 |
|
| Figure (A) shows normal red blood cells flowing freely through a blood vessel. The inset shows a cross-section of a normal red blood cell with normal haemoglobin. Figure (B) shows abnormal, sickled red blood cells sticking at the branching point in a blood vessel. The inset image shows a cross-section of a sickle cell with long polymerized sickle haemoglobin (HbS) strands stretching and distorting the cell shape to look like a crescent moon. | |
| Specialty | Hematology, medical genetics |
| Symptoms | Attacks of pain, anemia, swelling in the hands and feet, bacterial infections, stroke[1] |
| Complications | Chronic pain, stroke, aseptic bone necrosis, gallstones, leg ulcers, priapism, pulmonary hypertension, vision problems, kidney problems[2] |
| Usual onset | 5–6 months of age[1] |
| Causes | Genetic, Homozygous mutation in the hemoglobin S gene. [3] |
| Diagnostic method | Blood test[4] |
| Treatment | Vaccination, antibiotics, high fluid intake, folic acid supplementation, pain medication, blood transfusions[5][6] |
| Prognosis | Life expectancy 40–60 years (developed world)[2] |
| Frequency | 4.4 million (2015)[7] |
| Deaths | 114,800 (2015)[8] |
Sickle cell disease (SCD) is a group of blood disorders typically inherited.[2] The most common type is known as sickle cell anaemia.[2] It results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells.[2] This leads to a rigid, sickle-like shape under certain circumstances.[2] Problems in sickle cell disease typically begin around 5 to 6 months of age.[1] A number of health problems may develop, such as attacks of pain (known as a sickle cell crisis), anemia, swelling in the hands and feet, bacterial infections, and stroke.[1] Long-term pain may develop as people get older.[2] The average life expectancy in the developed world is 40 to 60 years.[2]
Sickle cell disease occurs when a person inherits two abnormal copies of the β-globin gene (HBB) that makes haemoglobin, one from each parent.[3] This gene occurs in chromosome 11.[9] Several subtypes exist, depending on the exact mutation in each haemoglobin gene.[2] An attack can be set off by temperature changes, stress, dehydration, and high altitude.[1] A person with a single abnormal copy does not usually have symptoms and is said to have sickle cell trait.[3] Such people are also referred to as carriers.[5] Diagnosis is by a blood test, and some countries test all babies at birth for the disease.[4] Diagnosis is also possible during pregnancy.[4]
The care of people with sickle cell disease may include infection prevention with vaccination and antibiotics, high fluid intake, folic acid supplementation, and pain medication.[5][6] Other measures may include blood transfusion and the medication hydroxycarbamide (hydroxyurea).[6] A small percentage of people can be cured by a transplant of bone marrow cells.[2]
As of 2015, about 4.4 million people have sickle cell disease, while an additional 43 million have sickle cell trait.[7][10] About 80% of sickle cell disease cases are believed to occur in Sub-Saharan Africa.[11] It also occurs to a lesser degree in parts of India, Southern Europe, West Asia, North Africa and among people of African origin (sub-Saharan) living in other parts of the world.[12] In 2015, it resulted in about 114,800 deaths.[8] The condition was first described in the medical literature by American physician James B. Herrick in 1910.[13][14] In 1949, its genetic transmission was determined by E. A. Beet and J. V. Neel.[14] In 1954, the protective effect against malaria of sickle cell trait was described.[14]
Signs and symptoms[edit]
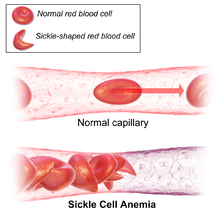
Sickle cells in human blood — both normal red blood cells and sickle-shaped cells are present.

Normal blood cells next to a sickle blood cell, coloured scanning electron microscope image
Signs of sickle cell disease usually begin in early childhood. The severity of symptoms can vary from person to person.[15] Sickle cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.[16]
Sickle cell crisis[edit]
The terms «sickle cell crisis» or «sickling crisis» may be used to describe several independent acute conditions occurring in patients with SCD, which results in anaemia and crises that could be of many types, including the vaso-occlusive crisis, aplastic crisis, splenic sequestration crisis, haemolytic crisis, and others. Most episodes of sickle cell crises last between five and seven days.[17] «Although infection, dehydration, and acidosis (all of which favor sickling) can act as triggers, in most instances, no predisposing cause is identified.»[18]
Vaso-occlusive crisis[edit]
The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis, and often organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration, analgesics, and blood transfusion; pain management requires opioid drug administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manages on nonsteroidal anti-inflammatory drugs such as diclofenac or naproxen. For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia devices are commonly used in this setting. Vaso-occlusive crisis involving organs such as the penis[19] or lungs are considered an emergency and treated with red blood cell transfusions. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.[20]
Splenic sequestration crisis[edit]
The spleen is frequently affected in sickle cell disease, as the sickle-shaped red blood cells cause narrowing of blood vessels and reduced function in clearing the defective cells.[21] It is usually infarcted before the end of childhood in individuals with sickle cell anaemia. This spleen damage increases the risk of infection from encapsulated organisms;[22][23] preventive antibiotics and vaccinations are recommended for those lacking proper spleen function.
Splenic sequestration crises are acute, painful enlargements of the spleen, caused by intrasplenic trapping of red cells and resulting in a precipitous fall in haemoglobin levels with the potential for hypovolemic shock. Sequestration crises are considered an emergency. If not treated, patients may die within 1–2 hours due to circulatory failure. Management is supportive, sometimes with blood transfusion. These crises are transient; they continue for 3–4 hours and may last for one day.[24]
Acute chest syndrome[edit]
Acute chest syndrome is defined by at least two of these signs or symptoms: chest pain, fever, pulmonary infiltrate or focal abnormality, respiratory symptoms, or hypoxemia.[20] It is the second-most common complication and it accounts for about 25% of deaths in patients with SCD. Most cases present with vaso-occlusive crises, and then develop acute chest syndrome.[25][26] Nevertheless, about 80% of people have vaso-occlusive crises during acute chest syndrome.[citation needed]
Aplastic crisis[edit]
Aplastic crises are instances of an acute worsening of the patient’s baseline anaemia, producing pale appearance, fast heart rate, and fatigue. This crisis is normally triggered by parvovirus B19, which directly affects production of red blood cells by invading the red cell precursors and multiplying in and destroying them.[27] Parvovirus infection almost completely prevents red blood cell production for two to three days. In normal individuals, this is of little consequence, but the shortened red cell life of SCD patients results in an abrupt, life-threatening situation. Reticulocyte counts drop dramatically during the disease (causing reticulocytopenia), and the rapid turnover of red cells leads to the drop in haemoglobin. This crisis takes 4 to 7 days to disappear. Most patients can be managed supportively; some need a blood transfusion.[28]
Haemolytic crisis[edit]
Haemolytic crises are acute accelerated drops in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in people with coexistent G6PD deficiency.[29] Another influence of hemolytic crises in sickle cell disease is oxidative stress on the erythrocytes, leukocytes, and platelets. When there is not enough red blood cell production in the bone marrow, the oxygen that the body receives, processes, and transports is unbalanced with the body’s antioxidants. There is an imbalance in the oxygen reactive species in the cells, which leads to more production of red blood cells that are not properly oxygenated or formed. Oxidative stress may lead to anemia because of the imbalance of oxygen in the tissue.[30]
Management is supportive, sometimes with blood transfusions.[20]
Other[edit]
One of the earliest clinical manifestations is dactylitis, presenting as early as six months of age, and may occur in children with sickle cell trait.[31] The crisis can last up to a month.[32] Given that pneumonia and sickling in the lung can both produce symptoms of acute chest syndrome, the patient is treated for both conditions.[33] It can be triggered by painful crisis, respiratory infection, bone-marrow embolisation, or possibly by atelectasis, opiate administration, or surgery.[34] Hematopoietic ulcers may also occur.[35]
Complications[edit]
Sickle cell anaemia can lead to various complications, including:
- Increased risk of severe bacterial infections is due to loss of functioning spleen tissue (and comparable to the risk of infections after having the spleen removed surgically). These infections are typically caused by encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae. Daily penicillin prophylaxis is the most commonly used treatment during childhood, with some haematologists continuing treatment indefinitely. Patients benefit today from routine vaccination for S. pneumoniae.[36]
- Stroke, which can result from a progressive narrowing of blood vessels, prevents oxygen from reaching the brain. Cerebral infarction occurs in children and cerebral haemorrhage in adults.[citation needed]
- Silent stroke causes no immediate symptoms, but is associated with damage to the brain. Silent stroke is probably five times as common as symptomatic stroke. About 10–15% of children with SCD have strokes, with silent strokes predominating in the younger patients.[37][38]
- Cholelithiasis (gallstones) and cholecystitis may result from excessive bilirubin production and precipitation due to prolonged haemolysis.[39]
- Avascular necrosis (aseptic bone necrosis) of the hip and other major joints may occur as a result of ischaemia.[40]
- Decreased immune reactions due to hyposplenism (malfunctioning of the spleen)[41]
- Priapism and infarction of the penis[42]
- Osteomyelitis (bacterial bone infection), the most common cause of osteomyelitis in SCD is Salmonella (especially the atypical serotypes Salmonella typhimurium, Salmonella enteritidis, Salmonella choleraesuis, and Salmonella paratyphi B), followed by Staphylococcus aureus and Gram-negative enteric bacilli perhaps because intravascular sickling of the bowel leads to patchy ischaemic infarction.[43]
- Acute papillary necrosis in the kidneys[citation needed]
- Leg ulcers[44]
- In eyes, background retinopathy, proliferative retinopathy, vitreous haemorrhages, and retinal detachments can result in blindness.[45] Regular annual eye checks are recommended.
- During pregnancy, intrauterine growth restriction, spontaneous abortion, and pre-eclampsia
- Chronic pain: Even in the absence of acute vaso-occlusive pain, many patients have unreported chronic pain.[46]
- Pulmonary hypertension (increased pressure on the pulmonary artery) can lead to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance, and episodes of syncope.[47] 21% of children and 30% of adults have evidence of pulmonary hypertension when tested; this is associated with reduced walking distance and increased mortality.[48]
- Cardiomyopathy and left ventricular diastolic dysfunction caused by fibrosis or scarring of cardiac tissues.[49][50] This also contributes to pulmonary hypertension, decreased exercise capacity, and arrhythmias.[51]
- Chronic kidney failure due to sickle-cell nephropathy manifests itself with hypertension, protein loss in the urine, loss of red blood cells in urine and worsened anaemia. If it progresses to end-stage kidney failure, it carries a poor prognosis.[52]
Genetics[edit]


Distribution of the sickle cell trait, shown in pink and purple
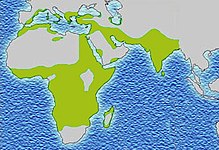
Historical distribution of malaria (no longer endemic in Europe), shown in green
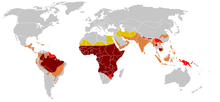
Modern distribution of malaria
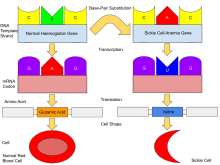
Base-pair substitution that causes sickle cell anemia
Normally, humans have haemoglobin A, which consists of two alpha and two beta chains, haemoglobin A2, which consists of two alpha and two delta chains, and haemoglobin F (HbF), consisting of two alpha and two gamma chains in their bodies. Of these three types, haemoglobin F dominates until about 6 weeks of age. Afterwards, haemoglobin A dominates throughout life.[53] In people diagnosed with sickle cell disease, at least one of the β-globin subunits in haemoglobin A is replaced with what is known as haemoglobin S. In sickle cell anaemia, a common form of sickle cell disease, haemoglobin S replaces both β-globin subunits in the haemoglobin.[15]
Sickle cell disease has an autosomal recessive pattern of inheritance from parents.[54] The types of haemoglobin a person makes in the red blood cells depend on what haemoglobin genes are inherited from her or his parents. If one parent has sickle cell anaemia and the other has sickle cell trait, then the child has a 50% chance of having sickle cell disease and a 50% chance of having sickle cell trait. When both parents have sickle cell trait, a child has a 25% chance of sickle cell disease; 25% do not carry any sickle cell alleles, and 50% have the heterozygous condition.[55]
Sickle cell gene mutation probably arose spontaneously in different geographic areas, as suggested by restriction endonuclease analysis. These variants are known as Cameroon, Senegal, Benin, Bantu, and Saudi-Asian. Their clinical importance is because some are associated with higher HbF levels, e.g., Senegal and Saudi-Asian variants, and tend to have milder disease.[56]
The gene defect is a single nucleotide mutation (see single-nucleotide polymorphism – SNP) (GAG codon changing to GTG) of the β-globin gene, which results in glutamate (E/Glu) being substituted by valine (V/Val) at position 6 (E6V substitution).[57][note 1] Haemoglobin S with this mutation is referred to as HbS, as opposed to the normal adult HbA. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structures of haemoglobin in conditions of normal oxygen concentration. However, under low oxygen concentration, HbS polymerizes and forms fibrous precipitates because the deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices (Phe 85, Leu 88).[58]
In people heterozygous for HbS (carriers of sickling haemoglobin), the polymerisation problems are minor because the normal allele can produce half of the haemoglobin. In people homozygous for HbS, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smooth, doughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated.[citation needed]
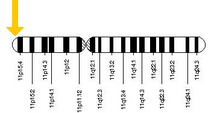
The allele responsible for sickle cell anaemia can be found on the short arm of chromosome 11, more specifically 11p15.5. A person who receives the defective gene from both father and mother develops the disease; a person who receives one defective and one healthy allele remains healthy, but can pass on the disease and is known as a carrier or heterozygote. Heterozygotes are still able to contract malaria, but their symptoms are generally less severe.[59]
Due to the adaptive advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India, and the Middle East.[60] Malaria was historically endemic to southern Europe, but it was declared eradicated in the mid-20th century, with the exception of rare sporadic cases.[61]
The malaria parasite has a complex lifecycle and spends part of it in red blood cells. In a carrier, the presence of the malaria parasite causes the red blood cells with defective haemoglobin to rupture prematurely, making the Plasmodium parasite unable to reproduce. Further, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people’s chances of survival actually increase if they carry sickle cell traits (selection for the heterozygote).[citation needed]
In the United States, with no endemic malaria, the prevalence of sickle cell anaemia among people of African ancestry is lower (about 0.25%) than among people in West Africa (about 4.0%) and is falling. Without endemic malaria, the sickle cell mutation is purely disadvantageous and tends to decline in the affected population by natural selection, and now artificially through prenatal genetic screening. However, the African American community descends from a significant admixture of several African and non-African ethnic groups and also represents the descendants of survivors of slavery and the slave trade. Thus, a degree of genetic dilution via crossbreeding with non-African people and high health-selective pressure through slavery (especially the slave trade and the frequently deadly Middle Passage) may be the most plausible explanations for the lower prevalence of sickle cell anaemia (and, possibly, other genetic diseases) among African Americans compared to West Africans. Another factor that limits the spread of sickle cell genes in North America is the relative absence of polygamy. In polygamous societies, affected males may father many children with multiple partners.[62]
Pathophysiology[edit]
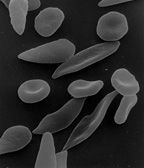
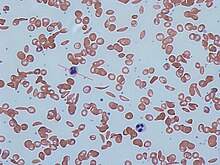
Scanning electron micrograph showing a mixture of red blood cells, some with round normal morphology, some with mild sickling showing elongation and bending
The loss of red blood cell elasticity is central to the pathophysiology of sickle cell disease. Normal red blood cells are quite elastic and have a biconcave disc shape, which allows the cells to deform to pass through capillaries.[63] In sickle cell disease, low oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell’s elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.[citation needed]
The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells, because of their shape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction.[64] Healthy red blood cells typically function for 90–120 days, but sickled cells only last 10–20 days.[65]
Diagnosis[edit]
In HbS, the complete blood count reveals haemoglobin levels in the range of 6–8 g/dl with a high reticulocyte count (as the bone marrow compensates for the destruction of sickled cells by producing more red blood cells). In other forms of sickle cell disease, Hb levels tend to be higher. A blood film may show features of hyposplenism (target cells and Howell-Jolly bodies).[66]
Sickling of the red blood cells, on a blood film, can be induced by the addition of sodium metabisulfite. The presence of sickle haemoglobin can also be demonstrated with the «sickle solubility test» (also called «sickledex»).[67] A mixture of haemoglobin S (HbS) in a reducing solution (such as sodium dithionite) gives a turbid appearance, whereas normal Hb gives a clear solution.[68]
Abnormal haemoglobin forms can be detected on haemoglobin electrophoresis, a form of gel electrophoresis on which the various types of haemoglobin move at varying speeds. Sickle cell haemoglobin (HgbS) and haemoglobin C with sickling (HgbSC)—the two most common forms—can be identified from there. The diagnosis can be confirmed with high-performance liquid chromatography. Genetic testing is rarely performed, as other investigations are highly specific for HbS and HbC.[69]
An acute sickle cell crisis is often precipitated by infection. Therefore, a urinalysis to detect an occult urinary tract infection, and chest X-ray to look for occult pneumonia should be routinely performed.[70]
People who are known carriers of the disease or at risk of having a child with sickle cell anemia may undergo genetic counseling. Genetic counselors work with families to discuss the benefits, limitations, and logistics of genetic testing options as well as the potential impact of testing and test results on the individual.[71][72] During pregnancy, genetic testing can be done on either a blood sample from the fetus or a sample of amniotic fluid. During the first trimester of pregnancy, chorionic villus sampling (CVS) is also a technique used for SCD prenatal diagnosis.[73] Since taking a blood sample from a fetus has greater risks, the latter test is usually used. Neonatal screening sometimes referred to as newborn screening, provides not only a method of early detection for individuals with sickle cell disease but also allows for the identification of the groups of people who carry the sickle cell trait.[74] Genetic counselors can help individuals of colour and their families tackle the racial and ethnic disparities that exist in healthcare.[75]
In 2010, there was significant consideration and debate in the US surrounding comprehensive screening of athletes for SCD.[76][77][78][79] The American Society of Hematology concluded in a statement in 2012 that they do not support testing or disclosure of sickle cell trait status as a prerequisite for participation in athletic activities due to lack of scientific evidence, inconsistency with good medical practice, and inconsistency with public health ethics. They recommended universal interventions to reduce exertion-related injuries and deaths effective for all athletes irrespective of their sickle cell status.[80]
Management[edit]
Treatment involves a number of measures. While it has been historically recommended that people with sickle cell disease avoid exercise, regular exercise may benefit people.[81] Dehydration should be avoided.[82] A diet high in calcium is recommended[83] but the effectiveness of vitamin D supplementation remains uncertain.[84] L-glutamine use was supported by the FDA starting at the age of five, as it decreases complications.[85]
Folic acid and penicillin[edit]
From birth to five years of age, penicillin daily, due to the immature immune system that makes them more prone to early childhood illnesses, is recommended.[86] Dietary supplementation of folic acid had been previously recommended by the WHO.[5] A 2016 Cochrane review of its use found «the effect of supplementation on anaemia and any symptoms of anaemia remains unclear» due to a lack of medical evidence.[87]
Malaria prevention[edit]

Possible advantage of being heterozygous for sickle cell anemia disease (A) vs. normal blood cell response (B) when infected with malaria
The protective effect of sickle cell trait does not apply to people with sickle cell disease; in fact, they are more vulnerable to malaria, since the most common cause of painful crises in malarial countries is infection with malaria. People with sickle cell disease living in malarial countries should receive lifelong medication for prevention.[88]
Vaso-occlusive crisis[edit]
Most people with sickle cell disease have intensely painful episodes called vaso-occlusive crises. However, the frequency, severity, and duration of these crises vary tremendously. Painful crises are treated symptomatically with pain medications; pain management requires opioid drug administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manages on NSAIDs (such as diclofenac or naproxen). For more severe crises, most patients require inpatient management for intravenous opioids.[89]
Extra fluids, administered either orally or intravenously, are a routine part of treatment of vaso-occlusive crises but the evidence about the most effective route, amount and type of fluid replacement remains uncertain.[90]
Crizanlizumab, a monoclonal antibody target towards p-selectin was approved in 2019 in the United States to reduce the frequency of vaso-occlusive crisis in those 16 years and older.[91]
Stroke prevention[edit]
Transcranial Doppler ultrasound (TCD) can detect children with sickle cell that have a high risk for stroke. The ultrasound test detects blood vessels partially obstructed by sickle cells by measuring the rate of blood into the brain, as blood flow velocity is inversely related to arterial diameter, and consequently, high blood-flow velocity is correlated with narrowing of the arteries.[92] In 2002 the National Institute of Health (NIH) issued a statement recommending that children with sickle cell get the Transcranial Doppler ultrasound screen annually, and in 2014 a panel of experts convened by the NIH issued guidelines reiterating the same recommendation. One review of medical records, by hematologist Dr. Julie Kanter at the University of Alabama at Birmingham, showed that on average only 48.4 percent of children with sickle cell get the recommended ultrasound test.[93]
A 1994 NIH study showed that children at risk for strokes who received blood transfusions had an annual stroke rate of less than 1 percent, whereas those children who did not receive blood transfusions had a 10 percent stroke rate per year. (Also see 1998 study in the New England Journal of Medicine.[92]) In addition to ultrasounds and blood transfusions, the inexpensive generic drug hydroxyurea can reduce the risk of irreversible organ and brain damage. Guidelines from NIH published in 2014 state that all children and adolescents should take hydroxyurea, as should adults with serious complications or three or more pain crises in a year.[94]
Acute chest syndrome[edit]
Management is similar to vaso-occlusive crisis, with the addition of antibiotics (usually a quinolone or macrolide, since cell wall-deficient [«atypical»] bacteria are thought to contribute to the syndrome),[95] oxygen supplementation for hypoxia, and close observation. In the absence of high quality evidence regarding the effectiveness of antibiotics for acute chest syndrome in people with sickle cell disease, there is no standard antibiotic treatment as of 2019.[96] It is recommended that people with suspected acute chest syndrome should be admitted to the hospital with worsening A-a gradient an indication for ICU admission.[20]
Should the pulmonary infiltrate worsen or the oxygen requirements increase, simple blood transfusion or exchange transfusion is indicated. The latter involves the exchange of a significant portion of the person’s red cell mass for normal red cells, which decreases the level of haemoglobin S in the patient’s blood. However, there is currently uncertain evidence about the possible benefits or harms of blood transfusion for acute chest syndrome in people with sickle cell disease.[97]
Hydroxyurea[edit]
Hydroxyurea, also known as hydroxycarbamide, probably reduces the frequency of painful episodes and the risk of life-threatening illness or death but there is currently insufficient evidence regarding the risk of adverse effects.[98] Hydroxyurea and phlebotomy combined may be more effective than transfusion and chelation combined in terms of pain, life-threatening illness and risk of death.[98]
It was the first approved drug for the treatment of sickle cell anaemia, and was shown to decrease the number and severity of attacks in 1995[99] and shown to possibly increase survival time in a study in 2003.[100] This is achieved, in part, by reactivating fetal haemoglobin production in place of the haemoglobin S that causes sickle cell anaemia. Hydroxyurea had previously been used as a chemotherapy agent, and some concern exists that long-term use may be harmful, but this risk is either absent or very small and the benefits likely outweigh the risks.[16][101]
Voxelotor was approved in the United States in 2019 to increase hemoglobin in people with SS disease.[102]
Blood transfusion[edit]
Blood transfusions are often used in the management of sickle cell disease in acute cases and to prevent complications by decreasing the number of red blood cells (RBCs) that can sickle by adding normal red blood cells.[103] In children, preventive RBC transfusion therapy has been shown to reduce the risk of first stroke or silent stroke when transcranial Doppler ultrasonography shows abnormal cerebral blood flow.[6] In those who have sustained a prior stroke event, it also reduces the risk of recurrent stroke and additional silent strokes.[104][105]
Bone marrow transplant[edit]
Bone marrow transplants have proven effective in children; they are the only known cure for SCD.[106] However, bone marrow transplants are difficult to obtain because of the specific HLA typing necessary. Ideally, a close relative (allogeneic) would donate the bone marrow necessary for transplantation. Some gene therapies are under development that would alter the patient’s own bone marrow stem cells ex vivo, which can then be transplanted back into the patient after chemotherapy eliminates the original unmodified cells.[107]
Avascular necrosis[edit]
When treating avascular necrosis of the bone in people with sickle cell disease, the aim of treatment is to reduce or stop the pain and maintain joint mobility.[40] Current treatment options include resting the joint, physical therapy, pain-relief medicine, joint-replacement surgery, or bone grafting.[40] High quality, randomized, controlled trials are needed to assess the most effective treatment option and determine if a combination of physical therapy and surgery is more effective than physical therapy alone.[40]
Psychological therapies[edit]
Psychological therapies such as patient education, cognitive therapy, behavioural therapy, and psychodynamic psychotherapy, that aim to complement current medical treatments, require further research to determine their effectiveness.[21]
Prognosis[edit]
About 90% of people survive to age 20, and close to 50% survive beyond age 50.[108] In 2001, according to one study performed in Jamaica, the estimated mean survival for people was 53 years for men and 58 years for women with homozygous SCD.[109] The specific life expectancy in much of the developing world is unknown.[110] In 1975 about 7.3% of people with SCD died before their 23rd birthday; while in 1989 2.6% of people with SCD died by the age of 20.[111]: 348
Epidemiology[edit]
The highest frequency of sickle cell disease is found in tropical regions, particularly sub-Saharan Africa, tribal regions of India, and the Middle East.[112] Migration of substantial populations from these high-prevalence areas to low-prevalence countries in Europe has dramatically increased in recent decades and in some European countries, sickle cell disease has now overtaken more familiar genetic conditions such as haemophilia and cystic fibrosis.[113] In 2015, it resulted in about 114,800 deaths.[8]
Sickle cell disease occurs more commonly among people whose ancestors lived in tropical and subtropical sub-Saharan regions where malaria is or was common. Where malaria is common, carrying a single sickle cell allele (trait) confers a heterozygote advantage; humans with one of the two alleles of sickle cell disease show less severe symptoms when infected with malaria.[114]
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.[115]
Africa[edit]
Three-quarters of sickle cell cases occur in Africa. A recent WHO report estimated that around 2% of newborns in Nigeria were affected by sickle cell anaemia, giving a total of 150,000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10 and 40% across equatorial Africa, decreasing to 1–2% on the North African coast and <1% in South Africa.[116]
Studies in Africa show a significant decrease in infant mortality rate, ages 2–16 months, because of the sickle cell trait. This happened in areas of predominant malarial cases.[117]
Uganda has the fifth-highest sickle cell disease burden in Africa.[118] One study indicates that 20 000 babies per year are born with sickle cell disease with the sickle cell trait at 13·3% and with disease 0·7%.[119]
| Country | Population(2020) | Subregion | % of Prevalence | Prevalence | Incidence |
| Angola | 32,866,272 | Middle Africa | 0.09375 | 3,081,213 | 14,869 |
| Cameroon | 26,545,863 | Middle Africa | 0.117 | 3,105,866 | 11,826 |
| DR Congo | 89,561,403 | Middle Africa | 0.1163333333 | 10,418,977 | 65,536 |
| Ghana | 31,072,940 | Western Africa | 0.09375 | 2,913,088 | 9,588 |
| Guinea | 13,132,795 | Western Africa | 0.139375 | 1,830,383 | 8,907 |
| Niger | 24,206,644 | Western Africa | 0.07025 | 1,700,517 | 8,756 |
| Nigeria | 206,139,589 | Western Africa | 0.1286666667 | 26,523,294 | 150,000 |
| Tanzania | 59,734,218 | Eastern Africa | 0.0545 | 3,255,515 | 19,585 |
| Uganda | 45,741,007 | Eastern Africa | 0.07025 | 3,213,306 | 17,936 |
| Zambia | 18,383,955 | Eastern Africa | 0.082 | 1,507,484 | 9,958 |
| Algeria | 43,851,044 | Northern Africa | 0.029 | 1,271,680 | 6,624 |
| Benin | 12,123,200 | Western Africa | 0.1286666667 | 1,559,852 | 8,125 |
| Botswana | 2,351,627 | Southern Africa | 0.029 | 68,197 | 355 |
| Burkina Faso | 20,903,273 | Western Africa | 0.07025 | 1,468,455 | 7,649 |
| Burundi | 11,890,784 | Eastern Africa | 0.023 | 273,488 | 1,425 |
| Cabo Verde | 555,987 | Western Africa | 0.023 | 12,788 | 67 |
| Central African Republic | 4,829,767 | Middle Africa | 0.082 | 396,041 | 2,063 |
| Chad | 16,425,864 | Middle Africa | 0.0585 | 960,913 | 5,005 |
| Comoros | 869,601 | Eastern Africa | 0.023 | 20,001 | 104 |
| Congo | 5,518,087 | Middle Africa | 0.1615 | 891,171 | 4,642 |
| Côte d’Ivoire | 26,378,274 | Western Africa | 0.07025 | 1,853,074 | 9,652 |
| Djibouti | 988,000 | Eastern Africa | 0.023 | 22,724 | 118 |
| Egypt | 102,334,404 | Northern Africa | 0.029 | 2,967,698 | 15,458 |
| Equatorial Guinea | 1,402,985 | Middle Africa | 0.181 | 253,940 | 1,323 |
| Eritrea | 3,546,421 | Eastern Africa | 0.023 | 81,568 | 425 |
| Eswatini | 1,160,164 | Southern Africa | 0.023 | 26,684 | 139 |
| Ethiopia | 114,963,588 | Eastern Africa | 0.029 | 3,333,944 | 17,366 |
| Gabon | 2,225,734 | Middle Africa | 0.181 | 402,858 | 2,098 |
| Gambia | 2,416,668 | Western Africa | 0.082 | 198,167 | 1,032 |
| Guinea-Bissau | 1,968,001 | Western Africa | 0.035 | 68,880 | 359 |
| Kenya | 53,771,296 | Eastern Africa | 0.04675 | 2,513,808 | 13,094 |
| Lesotho | 2,142,249 | Southern Africa | 0.023 | 49,272 | 257 |
| Liberia | 5,057,681 | Western Africa | 0.07025 | 355,302 | 1,851 |
| Libya | 6,871,292 | Northern Africa | 0.029 | 199,267 | 1,038 |
| Madagascar | 27,691,018 | Eastern Africa | 0.04675 | 1,294,555 | 6,743 |
| Malawi | 19,129,952 | Eastern Africa | 0.035 | 669,548 | 3,488 |
| Mali | 20,250,833 | Western Africa | 0.082 | 1,660,568 | 8,650 |
| Mauritania | 4,649,658 | Western Africa | 0.04675 | 217,372 | 1,132 |
| Mauritius | 1,271,768 | Eastern Africa | 0.023 | 29,251 | 152 |
| Morocco | 36,910,560 | Northern Africa | 0.029 | 1,070,406 | 5,576 |
| Mozambique | 31,255,435 | Eastern Africa | 0.035 | 1,093,940 | 5,698 |
| Namibia | 2,540,905 | Southern Africa | 0.03883333333 | 98,672 | 514 |
| Rwanda | 12,952,218 | Eastern Africa | 0.035 | 453,328 | 2,361 |
| São Tomé and Príncipe | 219,159 | Middle Africa | 0.181 | 39,668 | 207 |
| Senegal | 16,743,927 | Western Africa | 0.07025 | 1,176,261 | 6,127 |
| Seychelles | 98,347 | Eastern Africa | 0.023 | 2,262 | 12 |
| Sierra Leone | 7,976,983 | Western Africa | 0.1615 | 1,288,283 | 6,711 |
| Somalia | 15,893,222 | Eastern Africa | 0.029 | 460,903 | 2,401 |
| South Africa | 59,308,690 | Southern Africa | 0.029 | 1,719,952 | 8,959 |
| South Sudan | 11,193,725 | Eastern Africa | 0.04675 | 523,307 | 2,726 |
| Sudan | 43,849,260 | Northern Africa | 0.03883333333 | 1,702,813 | 8,870 |
| Togo | 8,278,724 | Western Africa | 0.09375 | 776,130 | 4,043 |
| Tunisia | 11,818,619 | Northern Africa | 0.023 | 271,828 | 1,416 |
| Zimbabwe | 14,862,924 | Eastern Africa | 0.035 | 520,202 | 2,710 |
| Total | 1,338,826,604 | Africa | 91,868,664 | 495,726 |
United States[edit]
The number of people with the disease in the United States is about 100,000 (one in 3,300), mostly affecting Americans of sub-Saharan African descent.[120] In the United States, about one out of 365 African-American children and one in every 16,300 Hispanic-American children have sickle cell anaemia.[121] The life expectancy for men with SCD is approximately 42 years of age while women live approximately six years longer.[122] An additional 2 million are carriers of the sickle cell trait.[123] Most infants with SCD born in the United States are identified by routine neonatal screening. As of 2016 all 50 states include screening for sickle cell disease as part of their newborn screen.[124] The newborn’s blood is sampled through a heel-prick and is sent to a lab for testing. The baby must have been eating for a minimum of 24 hours before the heel-prick test can be done. Some states also require a second blood test to be done when the baby is two weeks old to ensure the results.[125] Sickle cell anemia is the most common genetic disorder among African Americans. Approximately 8% are carriers and 1 in 375 are born with the disease.[126] Patient advocates for sickle cell disease have complained that it gets less government and private research funding than similar rare diseases such as cystic fibrosis, with researcher Elliott Vichinsky saying this shows racial discrimination or the role of wealth in health care advocacy.[127]
France[edit]

Percentage of newborns screened for sickle cell disease within Metropolitan France from 2006 to 2018
As a result of population growth in African-Caribbean regions of overseas France and immigration from North and sub-Saharan Africa to mainland France, sickle cell disease has become a major health problem in France.[128] SCD has become the most common genetic disease in the country, with an overall birth prevalence of one in 2,415 in metropolitan France, ahead of phenylketonuria (one in 10,862), congenital hypothyroidism (one in 3,132), congenital adrenal hyperplasia (one in 19,008) and cystic fibrosis (one in 5,014) for the same reference period.[citation needed]

Percentage of newborns screened regionally and overall for sickle cell disease in Metropolitan France in 2018
Since 2000, neonatal screening of SCD has been performed at the national level for all newborns defined as being «at-risk» for SCD based on ethnic origin (defined as those born to parents originating from sub-Saharan Africa, North Africa, the Mediterranean area (South Italy, Greece, and Turkey), the Arabic peninsula, the French overseas islands, and the Indian subcontinent).[129]
United Kingdom[edit]
In the United Kingdom, between 12,000 and 15,000 people are thought to have sickle cell disease [130] with an estimated 250,000 carriers of the condition in England alone. As the number of carriers is only estimated, all newborn babies in the UK receive a routine blood test to screen for the condition.[131] Due to many adults in high-risk groups not knowing if they are carriers, pregnant women and both partners in a couple are offered screening so they can get counselling if they have the sickle cell trait.[132] In addition, blood donors from those in high-risk groups are also screened to confirm whether they are carriers and whether their blood filters properly.[133] Donors who are found to be carriers are then informed and their blood, while often used for those of the same ethnic group, is not used for those with sickle cell disease who require a blood transfusion.[134]
West Asia[edit]
In Saudi Arabia, about 4.2% of the population carry the sickle cell trait and 0.26% have sickle cell disease. The highest prevalence is in the Eastern province, where approximately 17% of the population carry the gene and 1.2% have sickle cell disease.[135]
In 2005, Saudi Arabia introduced a mandatory premarital test including HB electrophoresis, which aimed to decrease the incidence of SCD and thalassemia.[136]
In Bahrain, a study published in 1998 that covered about 56,000 people in hospitals in Bahrain found that 2% of newborns have sickle cell disease, 18% of the surveyed people have the sickle cell trait, and 24% were carriers of the gene mutation causing the disease.[137] The country began screening of all pregnant women in 1992 and newborns started being tested if the mother was a carrier. In 2004, a law was passed requiring couples planning to marry to undergo free premarital counseling. These programs were accompanied by public education campaigns.[138]
India and Nepal[edit]
Sickle cell disease is common in some ethnic groups of central India,[139] where the prevalence has ranged from 9.4 to 22.2% in endemic areas of Madhya Pradesh, Rajasthan, and Chhattisgarh.[140] It is also endemic among Tharu people of Nepal and India; however, they have a sevenfold lower rate of malaria despite living in a malaria infested zone.[141]
Caribbean Islands[edit]
In Jamaica, 10% of the population carry the sickle cell gene, making it the most prevalent genetic disorder in the country.[142]
History[edit]
The first modern report of sickle cell disease may have been in 1846, where the autopsy of an executed runaway slave was discussed; the key finding was the absence of the spleen.[143][144] Reportedly, African slaves in the United States exhibited resistance to malaria, but were prone to leg ulcers.[144] The abnormal characteristics of the red blood cells, which later lent their name to the condition, was first described by Ernest E. Irons (1877–1959), intern to Chicago cardiologist and professor of medicine James B. Herrick (1861–1954), in 1910. Irons saw «peculiar elongated and sickle-shaped» cells in the blood of a man named Walter Clement Noel, a 20-year-old first-year dental student from Grenada. Noel had been admitted to the Chicago Presbyterian Hospital in December 1904 with anaemia.[13][145] Noel was readmitted several times over the next three years for «muscular rheumatism» and «bilious attacks» but completed his studies and returned to the capital of Grenada (St. George’s) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada.[13][14] Shortly after the report by Herrick, another case appeared in the Virginia Medical Semi-Monthly with the same title, «Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia.»[146] This article is based on a patient admitted to the University of Virginia Hospital on 15 November 1910.[147] In the later description by Verne Mason in 1922, the name «sickle cell anemia» is first used.[14][148] Childhood problems related to sickle cells disease were not reported until the 1930s, despite the fact that this cannot have been uncommon in African-American populations.[144]
Memphis physician Lemuel Diggs, a prolific researcher into sickle cell disease, first introduced the distinction between sickle cell disease and trait in 1933, although until 1949, the genetic characteristics had not been elucidated by James V. Neel and E.A. Beet.[14] 1949 was the year when Linus Pauling described the unusual chemical behaviour of haemoglobin S, and attributed this to an abnormality in the molecule itself.[14][149] The molecular change in HbS was described in 1956 by Vernon Ingram.[150] The late 1940s and early 1950s saw further understanding in the link between malaria and sickle cell disease. In 1954, the introduction of haemoglobin electrophoresis allowed the discovery of particular subtypes, such as HbSC disease.[14]
Large-scale natural history studies and further intervention studies were introduced in the 1970s and 1980s, leading to widespread use of prophylaxis against pneumococcal infections amongst other interventions. Bill Cosby’s Emmy-winning 1972 TV movie, To All My Friends on Shore, depicted the story of the parents of a child with sickle cell disease.[151] The 1990s had the development of hydroxycarbamide, and reports of cure through bone marrow transplantation appeared in 2007.[14]
Some old texts refer to it as drepanocytosis.[152]
Society and culture[edit]
United States[edit]
Effective 15 September 2017, the U.S. Social Security Administration issued a Policy Interpretation Ruling providing background information on sickle cell disease and a description of how Social Security evaluates the disease during its adjudication process for disability claims.[153][154]
In the U.S., there are stigmas surrounding SCD that discourage people with SCD from receiving necessary care. These stigmas mainly affect people of African American and Latin American ancestries, according to the National Heart, Lung, and Blood Institute.[155] People with SCD experience the impact of stigmas of the disease on multiple aspects of life including social and psychological well-being. Studies have shown that those with SCD frequently feel as though they must keep their diagnosis a secret to avoid discrimination in the workplace and also among peers in relationships.[156] In the 1960s, the US government supported initiatives for workplace screening for genetic diseases in an attempt to be protective towards people with SCD. By having this screening, it was intended that employees would not be placed in environments that could potentially be harmful and trigger SCD.[157]
Uganda[edit]
Uganda has the 5th highest sickle cell disease (SCD) burden in the world.[158] In Uganda, social stigma exists for those with sickle cell disease because of the lack of general knowledge of the disease. The general gap in knowledge surrounding sickle cell disease is noted among adolescents and young adults due to the culturally sanctioned secrecy about the disease.[158] While most people have heard generally about the disease, a large portion of the population is relatively misinformed about how SCD is diagnosed or inherited. Those who are informed about the disease learned about it from family or friends and not from health professionals. Failure to provide the public with information about sickle cell disease results in a population with a poor understanding of the causes of the disease, symptoms, and prevention techniques.[159] The differences, physically and socially, that arise in those with sickle cell disease, such as jaundice, stunted physical growth, and delayed sexual maturity, can also lead them to become targets of bullying, rejection, and stigma.[158]
Rate of sickle cell disease in Uganda[edit]
The data compiled on sickle cell disease in Uganda has not been updated since the early 1970s. The deficiency of data is due to a lack of government research funds, even though Ugandans die daily from SCD.[160] Data shows that the trait frequency of sickle cell disease is 20% of the population in Uganda.[160] This means that 66 million people are at risk of having a child who has sickle cell disease.[160] It is also estimated that about 25,000 Ugandans are born each year with SCD and 80% of those people don’t live past five years old.[160] SCD also contributes 25% to the child mortality rate in Uganda.[160] The Bamba people of Uganda, located in the southwest of the country, carry 45% of the gene which is the highest trait frequency recorded in the world.[160] The Sickle Cell Clinic in Mulago is only one sickle cell disease clinic in the country and on average sees 200 patients a day.[160]
Misconceptions about sickle cell disease[edit]
The stigma around the disease is particularly bad in regions of the country that are not as affected. For example, Eastern Ugandans tend to be more knowledgeable of the disease than Western Ugandans, who are more likely to believe that sickle cell disease resulted as a punishment from God or witchcraft.[161] Other misconceptions about SCD include the belief that it is caused by environmental factors but, in reality, SCD is a genetic disease.[162] There have been efforts throughout Uganda to address the social misconceptions about the disease. In 2013, the Uganda Sickle Cell Rescue Foundation was established to spread awareness of sickle cell disease and combat the social stigma attached to the disease.[163] In addition to this organization’s efforts, there is a need for the inclusion of sickle cell disease education in preexisting community health education programs in order to reduce the stigmatization of sickle cell disease in Uganda.[159]
[edit]
The deeply rooted stigma of SCD from society causes families to often hide their family members’ sick status for fear of being labeled, cursed, or left out of social events.[164] Sometimes in Uganda, when it is confirmed that a family member has sickle cell disease, intimate relationships with all members of the family are avoided.[164] The stigmatization and social isolation people with sickle cell disease tend to experience is often the consequence of popular misconceptions that people with SCD should not socialize with those free from the disease. This mentality robs people with SCD of the right to freely participate in community activities like everyone else[158] SCD-related stigma and social isolation in schools, especially, can make a life for young people living with sickle cell disease extremely difficult.[158] For school-aged children living with SCD, the stigma they face can lead to peer rejection.[158] Peer rejection involves the exclusion from social groups or gatherings. It often leads the excluded individual to experience emotional distress and may result in their academic underperformance, avoidance of school, and occupational failure later in life.[158] This social isolation is also likely to negatively impact people with SCD’s self-esteem and overall quality of life.[158]
Mothers of children with sickle cell disease tend to receive disproportionate amounts of stigma from their peers and family members. These women will often be blamed for their child’s diagnosis of SCD, especially if SCD is not present in earlier generations, due to the suspicion that the child’s poor health may have been caused by the mother’s failure to implement preventative health measures or promote a healthy environment for her child to thrive.[162] The reliance on theories related to environmental factors to place blame on the mother reflects many Ugandan’s poor knowledge of how the disease is acquired as it is determined by genetics, not environment.[162] Mothers of children with sickle cell disease are also often left with very limited resources to safeguard their futures against the stigma of having SCD.[162] This lack of access to resources results from their subordinating roles within familial structures as well as the class disparities that hinder many mothers’ ability to satisfy additional childcare costs and responsibilities.[162]
Women living with SCD who become pregnant often face extreme discrimination and discouragement in Uganda. These women are frequently branded by their peers as irresponsible for having a baby while living with sickle cell disease or even engaging in sex while living with SCD.[citation needed] The criticism and judgement these women receive, not only from healthcare professionals but also from their families, often leaves them feeling alone, depressed, anxious, ashamed, and with very little social support.[citation needed] Most pregnant women with SCD also go on to be single mothers as it is common for them to be left by their male partners who claim they were unaware of their partner’s SCD status.[citation needed] Not only does the abandonment experienced by these women cause emotional distress for them, but this low level of parental support can be linked to depressive symptoms and overall lower quality of life for the child once they are born.[165]
United Kingdom[edit]
In 2021 many patients were found to be afraid to visit hospitals so purchasing pain relief to treat themselves outside the NHS, such was the level of ignorance among staff. They were often waiting a long time for pain relief, and sometimes suspected of “drugs-seeking” behaviour. Delays to treatment, failure to inform the hospital haematology team and poor pain management had caused deaths. Specialist haematology staff prefer to work in bigger, teaching hospitals, leading to shortages of expertise elsewhere.[166] In 2021 the NHS initiated its first new treatment in 20 years for Sickle Cell. This involved the use of Crizanlizumab, a drug given via transfusion drips, which reduces the number of visits to A and E by sufferers. The treatment can be accessed, via consultants, at any of ten new hubs set up around the country.[167] In the same year, however, an All-Party Parliamentary Group produced a report on Sickle Cell and Thalassaemia entitled ‘No-one is listening’.[168] Partly in response to this, on 19 June 2022, World Sickle Cell Day, the NHS launched a campaign called » Can you tell it’s sickle cell?». The campaign had twin aims. One was to increase awareness of the key signs and symptoms of the blood disorder so that people are as alert to signs of a sickle cell crisis as they are to an imminent heart attack or stroke. The second aim was to set up a new training programme to help paramedics, Accident and Emergency staff, carers and the general public to care effectively for sufferers in crisis.[169]
Research[edit]
Umbilical cord blood transplant[edit]
While umbilical cord blood transplant can potentially cure the condition, a suitable donor is available in only 10% of people.[170] About 7% of people also die as a result of the procedure and graft versus host disease may occur.[170]
Gene therapy[edit]
Diseases such as sickle cell disease for which a person’s normal phenotype or cell function may be restored in cells that have the disease by a normal copy of the gene that is mutated, may be a good candidate for gene therapy treatment. The risks and benefits related to gene therapy for sickle cell disease are not known.[171]
In 2001, sickle cell disease reportedly had been successfully treated in mice using gene therapy.[172][173] The researchers used a viral vector to make the mice—which have essentially the same defect that causes human sickle cell disease—express production of fetal haemoglobin (HbF), which an individual normally ceases to produce shortly after birth. In humans, using hydroxyurea to stimulate the production of HbF has been known to temporarily alleviate sickle cell disease symptoms. The researchers demonstrated that this gene therapy method is a more permanent way to increase therapeutic HbF production.[174]
Phase 1 clinical trials of gene therapy for sickle cell disease in humans were started in 2014. The clinical trials will assess the safety of lentiviral vector-modified bone marrow for adults with severe sickle cell disease.[175][176] As of 2020, however, no randomized controlled trials have been reported.[171] A case report for the first person treated was published in March 2017, with a few more people being treated since then.[177][178]
Gene editing platforms like CRISPR/Cas9 have been used to correct the disease-causing mutation in hematopoietic stem cells taken from a person with the condition.[179] In July 2019 the gene-editing tool CRISPR was used to edit bone marrow cells from a person with SCD to boost fetal haemoglobin by inhibiting the BCL11A gene.[180][181] A number of researchers have considered the ethical implications of SCD being one of the first potential applications of CRISPR technology, given the historical abuses and neglect of the African American community by the medical field.[182]
In 2017 twelve clinical trials were focusing on gene therapy to treat sickle cell anemia. Of those 12 trials, four of them replaced the mutated HBB gene with a healthy one. Three trials used Mozobil, a medication used to treat types of cancer, to determine whether the increase of stem cells can be used for gene therapy. One trial focused on analyzing bone marrow samples from patients with sickle cell anemia. Another trial experimented with using umbilical cord blood from babies both with and without sickle cell anemia to develop gene therapy.[183]
Hematopoietic stem cell transplantation[edit]
There is no strong medical evidence to determine the risks and potential benefits related to treating people with sickle cell disease with hematopoietic stem cell transplantations.[184]
Notes[edit]
- ^ Historic numbering put this glutamic acid residue at position 6 due to skipping the methionine (M/Met) start codon in protein amino acid position numbering. Current nomenclature calls for counting the methionine as the first amino acid, resulting in the glutamic acid residue falling at position 7. Many references still refer to position 6 and both should likely be referenced for clarity.
References[edit]
- ^ a b c d e «What Are the Signs and Symptoms of Sickle Cell Disease?». National Heart, Lung, and Blood Institute. 12 June 2015. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- ^ a b c d e f g h i j «What Is Sickle Cell Disease?». National Heart, Lung, and Blood Institute. 12 June 2015. Archived from the original on 6 March 2016. Retrieved 8 March 2016.
- ^ a b c «What Causes Sickle Cell Disease?». National Heart, Lung, and Blood Institute. 12 June 2015. Archived from the original on 24 March 2016. Retrieved 8 March 2016.
- ^ a b c «How Is Sickle Cell Disease Diagnosed?». National Heart, Lung, and Blood Institute. 12 June 2015. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- ^ a b c d «Sickle-cell disease and other haemoglobin disorders Fact sheet N°308». January 2011. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- ^ a b c d «How Is Sickle Cell Disease Treated?». National Heart, Lung, and Blood Institute. 12 June 2015. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- ^ a b Allen C, Arora M, Barber RM, Bhutta ZA, Brown A, Carter A, et al. (GBD 2015 Disease and Injury Incidence and Prevalence Collaborators) (October 2016). «Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015». Lancet. 388 (10053): 1545–1602. doi:10.1016/S0140-6736(16)31678-6. PMC 5055577. PMID 27733282.
- ^ a b c Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A, et al. (GBD 2015 Mortality and Causes of Death Collaborators) (October 2016). «Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015». Lancet. 388 (10053): 1459–1544. doi:10.1016/S0140-6736(16)31012-1. PMC 5388903. PMID 27733281.
- ^ «Learning About Sickle Cell Disease». National Human Genome Research Institute. 9 May 2016. Archived from the original on 4 January 2017. Retrieved 23 January 2017.
- ^ Vos T, Barber RM, Bell B, Bertozzi-Villa A, Biryukov S, Bolliger I, et al. (Global Burden of Disease Study 2013 Collaborators) (August 2015). «Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013». Lancet. 386 (9995): 743–800. doi:10.1016/s0140-6736(15)60692-4. PMC 4561509. PMID 26063472.
- ^ Rees DC, Williams TN, Gladwin MT (December 2010). «Sickle-cell disease». Lancet. 376 (9757): 2018–2031. doi:10.1016/s0140-6736(10)61029-x. PMID 21131035. S2CID 29909566.
- ^ Elzouki AY (2012). Textbook of clinical pediatrics (2 ed.). Berlin: Springer. p. 2950. ISBN 9783642022012.
- ^ a b c Savitt TL, Goldberg MF (January 1989). «Herrick’s 1910 case report of sickle cell anemia. The rest of the story». JAMA. 261 (2): 266–271. doi:10.1001/jama.261.2.266. PMID 2642320.
- ^ a b c d e f g h i Serjeant GR (December 2010). «One hundred years of sickle cell disease». British Journal of Haematology. 151 (5): 425–429. doi:10.1111/j.1365-2141.2010.08419.x. PMID 20955412.
- ^ a b «Sickle cell disease: MedlinePlus Genetics». medlineplus.gov. Retrieved 22 October 2022.
- ^ a b Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. (September 2014). «Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members». JAMA. 312 (10): 1033–1048. doi:10.1001/jama.2014.10517. PMID 25203083. S2CID 37681044.
- ^ «BestBets: How long should an average sickle cell crisis last?». Archived from the original on 17 June 2010. Retrieved 27 November 2010.
- ^ Kumar V, Abbas AK, Fausto N, Aster J (28 May 2009). Robbins and Cotran Pathologic Basis of Disease (Professional Edition: Expert Consult – Online (Robbins Pathology) ed.). Elsevier Health. pp. Kindle Locations 33498–33499.
- ^ Olujohungbe A, Burnett AL (March 2013). «How I manage priapism due to sickle cell disease». British Journal of Haematology. 160 (6): 754–765. doi:10.1111/bjh.12199. PMID 23293942.
- ^ a b c d Glassberg J (August 2011). «Evidence-based management of sickle cell disease in the emergency department». Emergency Medicine Practice. 13 (8): 1–20, quiz 20. PMID 22164362.
- ^ a b Anie KA, Green J (May 2015). «Psychological therapies for sickle cell disease and pain». The Cochrane Database of Systematic Reviews. 2015 (5): CD001916. doi:10.1002/14651858.CD001916.pub3. PMC 7063720. PMID 25966336.
- ^ Pearson HA (August 1977). «Sickle cell anemia and severe infections due to encapsulated bacteria» (Free full text). The Journal of Infectious Diseases. 136 (Suppl): S25–S30. doi:10.1093/infdis/136.Supplement.S25. PMID 330779. Archived from the original on 27 May 2016.
- ^ Wong WY, Powars DR, Chan L, Hiti A, Johnson C, Overturf G (March 1992). «Polysaccharide encapsulated bacterial infection in sickle cell anemia: a thirty year epidemiologic experience». American Journal of Hematology. 39 (3): 176–182. doi:10.1002/ajh.2830390305. PMID 1546714. S2CID 19977178.
- ^ Khatib R, Rabah R, Sarnaik SA (January 2009). «The spleen in the sickling disorders: an update». Pediatric Radiology. 39 (1): 17–22. doi:10.1007/s00247-008-1049-9. PMID 19002450. S2CID 2547649.
- ^ Mekontso Dessap A, Leon R, Habibi A, Nzouakou R, Roudot-Thoraval F, Adnot S, et al. (March 2008). «Pulmonary hypertension and cor pulmonale during severe acute chest syndrome in sickle cell disease». American Journal of Respiratory and Critical Care Medicine. 177 (6): 646–653. CiteSeerX 10.1.1.504.790. doi:10.1164/rccm.200710-1606OC. PMID 18174543.
- ^ Paul RN, Castro OL, Aggarwal A, Oneal PA (September 2011). «Acute chest syndrome: sickle cell disease». European Journal of Haematology. 87 (3): 191–207. doi:10.1111/j.1600-0609.2011.01647.x. PMID 21615795. S2CID 40320701.
- ^ Kumar V, Abbas AK, Fausto N, Aster J (28 May 2009). Robbins and Cotran Pathologic Basis of Disease (Professional Edition: Expert Consult – Online (Robbins Pathology) ed.). Elsevier Health. pp. Kindle Location 33329.
- ^ Slavov SN, Kashima S, Pinto AC, Covas DT (August 2011). «Human parvovirus B19: general considerations and impact on patients with sickle-cell disease and thalassemia and on blood transfusions». FEMS Immunology and Medical Microbiology. 62 (3): 247–262. doi:10.1111/j.1574-695X.2011.00819.x. PMID 21585562.
- ^ Balgir RS (March 2012). «Community expansion and gene geography of sickle cell trait and G6PD deficiency, and natural selection against malaria: experience from tribal land of India». Cardiovascular & Hematological Agents in Medicinal Chemistry. 10 (1): 3–13. doi:10.2174/187152512799201190. PMID 22264009.
- ^ Fibach, Eitan; Rachmilewitz, Eliezer (2008). «The Role of Oxidative Stress in Hemolytic Anemia». Current Molecular Medicine. 8 (7): 609–619. doi:10.2174/156652408786241384. PMID 18991647.
- ^ Jadavji T, Prober CG (April 1985). «Dactylitis in a child with sickle cell trait». Canadian Medical Association Journal. 132 (7): 814–815. PMC 1345873. PMID 3978504.
- ^ Worrall VT, Butera V (December 1976). «Sickle-cell dactylitis». The Journal of Bone and Joint Surgery. American Volume. 58 (8): 1161–1163. doi:10.2106/00004623-197658080-00024. PMID 1002763. Archived from the original on 23 September 2016.
- ^ Miller ST (May 2011). «How I treat acute chest syndrome in children with sickle cell disease». Blood. 117 (20): 5297–5305. doi:10.1182/blood-2010-11-261834. PMID 21406723. S2CID 206896811.
- ^ Friend, A.; Girzadas, D. (2021). «Acute Chest Syndrome». StatPearls. StatPearls Publishing. PMID 28722902. Retrieved 21 March 2021.
- ^ James WD, Berger TG, et al. (2006). Andrews’ Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 847. ISBN 978-0-7216-2921-6.
- ^ Kavanagh PL, Sprinz PG, Vinci SR, Bauchner H, Wang CJ (December 2011). «Management of children with sickle cell disease: a comprehensive review of the literature». Pediatrics. 128 (6): e1552–e1574. doi:10.1542/peds.2010-3686. PMID 22123880. S2CID 14524078. Archived from the original on 4 March 2016.
- ^ Adams RJ, Ohene-Frempong K, Wang W (2001). «Sickle cell and the brain». Hematology. American Society of Hematology. Education Program. 2001 (1): 31–46. doi:10.1182/asheducation-2001.1.31. PMID 11722977.
- ^ Adams RJ (November 2007). «Big strokes in small persons». Archives of Neurology. 64 (11): 1567–1574. doi:10.1001/archneur.64.11.1567. PMID 17998439.
- ^ «Cholelithiasis». The Lecturio Medical Concept Library. Retrieved 25 August 2021.
- ^ a b c d Martí-Carvajal AJ, Solà I, Agreda-Pérez LH (December 2019). «Treatment for avascular necrosis of bone in people with sickle cell disease». The Cochrane Database of Systematic Reviews. 2019 (12): CD004344. doi:10.1002/14651858.CD004344.pub7. PMC 6894369. PMID 31803937.
- ^ Kenny MW, George AJ, Stuart J (July 1980). «Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism». Journal of Clinical Pathology. 33 (7): 622–625. doi:10.1136/jcp.33.7.622. PMC 1146172. PMID 7430367.
- ^ Chrouser KL, Ajiboye OB, Oyetunji TA, Chang DC (April 2011). «Priapism in the United States: the changing role of sickle cell disease». American Journal of Surgery. 201 (4): 468–474. doi:10.1016/j.amjsurg.2010.03.017. PMID 21421100.
- ^ Almeida A, Roberts I (May 2005). «Bone involvement in sickle cell disease». British Journal of Haematology. 129 (4): 482–490. doi:10.1111/j.1365-2141.2005.05476.x. PMID 15877730. S2CID 908481. Archived from the original on 16 December 2012.
- ^ Rudge FW (1991). «Hyperbaric oxygen therapy in the treatment of sickle cell leg ulcers». J. Hyperbaric Med. 6 (1): 1–4. Archived from the original on 15 April 2013. Retrieved 23 March 2011.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Elagouz M, Jyothi S, Gupta B, Sivaprasad S (July 2010). «Sickle cell disease and the eye: old and new concepts». Survey of Ophthalmology. 55 (4): 359–377. doi:10.1016/j.survophthal.2009.11.004. PMID 20452638.
- ^ Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, et al. (January 2008). «Daily assessment of pain in adults with sickle cell disease». Annals of Internal Medicine. 148 (2): 94–101. CiteSeerX 10.1.1.690.5870. doi:10.7326/0003-4819-148-2-200801150-00004. PMID 18195334. S2CID 34924760.
- ^ Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT (June 2014). «Pulmonary arterial hypertension: the clinical syndrome». Circulation Research. 115 (1): 115–130. doi:10.1161/CIRCRESAHA.115.301146. PMC 4096686. PMID 24951762.
- ^ Caughey MC, Poole C, Ataga KI, Hinderliter AL (August 2015). «Estimated pulmonary artery systolic pressure and sickle cell disease: a meta-analysis and systematic review». British Journal of Haematology. 170 (3): 416–424. doi:10.1111/bjh.13447. PMID 25854714. S2CID 23920740.
- ^ Niss O, Quinn CT, Lane A, Daily J, Khoury PR, Bakeer N, et al. (March 2016). «Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease». JACC. Cardiovascular Imaging. 9 (3): 243–252. doi:10.1016/j.jcmg.2015.05.013. PMC 4788530. PMID 26897687.
- ^ Niss O, Fleck R, Makue F, Alsaied T, Desai P, Towbin JA, et al. (July 2017). «Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia». Blood. 130 (2): 205–213. doi:10.1182/blood-2017-02-767624. PMC 5510791. PMID 28507082.
- ^ Rai P, Niss O, Malik P (November 2017). «A reappraisal of the mechanisms underlying the cardiac complications of sickle cell anemia». Pediatric Blood & Cancer. 64 (11): e26607. doi:10.1002/pbc.26607. PMID 28453224. S2CID 24444332.
- ^ Powars DR, Elliott-Mills DD, Chan L, Niland J, Hiti AL, Opas LM, Johnson C (October 1991). «Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality». Annals of Internal Medicine. 115 (8): 614–620. doi:10.7326/0003-4819-115-8-614. PMID 1892333.
- ^ Sankaran VG, Orkin SH (January 2013). «The switch from fetal to adult hemoglobin». Cold Spring Harbor Perspectives in Medicine. 3 (1): a011643. doi:10.1101/cshperspect.a011643. PMC 3530042. PMID 23209159.
- ^ «Sickle Cell Disease». NORD (National Organization for Rare Disorders). Retrieved 10 June 2019.
- ^ «sickle cell disease». Genetics Home Reference. Archived from the original on 15 May 2016. Retrieved 7 May 2016.
- ^ Green NS, Fabry ME, Kaptue-Noche L, Nagel RL (October 1993). «Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia». American Journal of Hematology. 44 (2): 145–146. doi:10.1002/ajh.2830440214. PMID 7505527. S2CID 27341091.
- ^ Clancy S (2008). «Genetic mutation». Nature Education. 1 (1): 187.
- ^ Odièvre MH, Verger E, Silva-Pinto AC, Elion J (October 2011). «Pathophysiological insights in sickle cell disease». The Indian Journal of Medical Research. 134 (1): 532–537. doi:10.1007/bf00168807. PMC 3237253. PMID 22089617.
- ^ Allison AC (October 2009). «Genetic control of resistance to human malaria». Current Opinion in Immunology. 21 (5): 499–505. doi:10.1016/j.coi.2009.04.001. PMID 19442502.
- ^ Kwiatkowski DP (August 2005). «How malaria has affected the human genome and what human genetics can teach us about malaria». American Journal of Human Genetics. 77 (2): 171–192. doi:10.1086/432519. PMC 1224522. PMID 16001361.
- ^ Ponçon N, Toty C, L’Ambert G, Le Goff G, Brengues C, Schaffner F, Fontenille D (February 2007). «Biology and dynamics of potential malaria vectors in Southern France». Malaria Journal. 6 (1): 18. doi:10.1186/1475-2875-6-18. PMC 1808464. PMID 17313664.
- ^ Lesi FE, Bassey EE (July 1972). «Family study in sickle cell disease in Nigeria». Journal of Biosocial Science. 4 (3): 307–313. doi:10.1017/S0021932000008622. PMID 5041262. S2CID 41719342.
- ^ Capriotti T, Frizzell JP (2016). Pathophysiology : introductory concepts and clinical perspectives. Philadelphia. ISBN 9780803615717. OCLC 900626405.
- ^ «How Does Sickle Cell Cause Disease?». Archived from the original on 23 September 2010. Retrieved 27 November 2010.
- ^ «Sickle Cell Anemia: eMedicine Emergency Medicine». Archived from the original on 4 December 2010. Retrieved 27 November 2010.
- ^ Kruzliak, Peter (19 July 2012), «Hematologic manifestations of celiac disease», Celiac Disease – From Pathophysiology to Advanced Therapies, InTech, doi:10.5772/31233, ISBN 978-953-51-0684-5
- ^ Atkinson K, Mabey D (23 May 2019). Revolutionizing Tropical Medicine: Point-of-Care Tests, New Imaging Technologies and Digital Health. Wiley. p. 227. ISBN 978-1-119-28265-5.
- ^ McPherson RA, Pincus MR (2017). Henry’s Clinical Diagnosis and Management by Laboratory Methods (23 ed.). Elsevier Health Sciences. p. 578. ISBN 978-0-323-41315-2.
- ^ Clarke GM, Higgins TN (August 2000). «Laboratory investigation of hemoglobinopathies and thalassemias: review and update». Clinical Chemistry. 46 (8 Pt 2): 1284–1290. doi:10.1093/clinchem/46.8.1284. PMID 10926923. Archived from the original on 20 March 2008.
- ^ «BestBets: Does routine urinalysis and chest radiography detect occult bacterial infection in sickle cell patients presenting to the accident and emergency department with painful crisis?». Archived from the original on 17 June 2010. Retrieved 27 November 2010.
- ^ «National Society of Genetic Counselors : About Genetic Counselors». www.nsgc.org. Retrieved 24 February 2021.
- ^ «ABGC – Information for Certified Genetic Counselors | ABGC». www.abgc.net. Retrieved 23 February 2021.
- ^ Colah, R. B., Gorakshakar, A. C., & Nadkarni, A. H. (2011). Invasive & non-invasive approaches for prenatal diagnosis of haemoglobinopathies: experiences from India. The Indian Journal of Medical Research, 134(4), 552–560.
- ^ Lee, C., Davies, S.,& Dezatoux, C. (2000). Neonatal Screening for sickle cell disease. The Cochrane Collaboration. John Wiley & Sons, Ltd.
- ^ Stallings E (27 July 2019). «Genetic Counselors Of Color Tackle Racial, Ethnic Disparities In Health Care». NPR. Retrieved 23 February 2021.
- ^ Goldsmith JC, Bonham VL, Joiner CH, Kato GJ, Noonan AS, Steinberg MH (March 2012). «Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications». American Journal of Hematology. 87 (3): 340–346. doi:10.1002/ajh.22271. PMC 3513289. PMID 22307997.
- ^ Bonham VL, Dover GJ, Brody LC (September 2010). «Screening student athletes for sickle cell trait—a social and clinical experiment». The New England Journal of Medicine. 363 (11): 997–999. doi:10.1056/NEJMp1007639. PMID 20825310.
- ^ Acharya K, Benjamin HJ, Clayton EW, Ross LF (November 2011). «Attitudes and beliefs of sports medicine providers to sickle cell trait screening of student athletes». Clinical Journal of Sport Medicine. 21 (6): 480–485. doi:10.1097/JSM.0b013e31822e8634. PMID 21959797. S2CID 11404187.
- ^ Ferrari R, Parker LS, Grubs RE, Krishnamurti L (December 2015). «Sickle Cell Trait Screening of Collegiate Athletes: Ethical Reasons for Program Reform». Journal of Genetic Counseling. 24 (6): 873–877. doi:10.1007/s10897-015-9849-1. PMID 26040250. S2CID 15889144.
- ^ «Statement on Screening for Sickle Cell Trait and Athletic Participation». www.hematology.org. Retrieved 24 February 2021.
- ^ Martin C, Pialoux V, Faes C, Charrin E, Skinner S, Connes P (February 2018). «Does physical activity increase or decrease the risk of sickle cell disease complications?». British Journal of Sports Medicine. 52 (4): 214–218. doi:10.1136/bjsports-2015-095317. PMID 26701924. S2CID 24464344.
- ^ «Keeping Well with Sickle Cell Disease — Brent Sickle Cell & Thalassaemia Centre». www.sickle-thal.nwlh.nhs.uk. Archived from the original on 3 October 2019. Retrieved 4 October 2019.
- ^ «Nutrition for the Child with Sickle Cell Anemia». www.eatright.org. Archived from the original on 19 June 2020. Retrieved 5 October 2019.
- ^ Soe HH, Abas AB, Than NN, Ni H, Singh J, Said AR, Osunkwo I (May 2020). «Vitamin D supplementation for sickle cell disease». The Cochrane Database of Systematic Reviews. 5 (9): CD010858. doi:10.1002/14651858.CD010858.pub3. PMC 7386793. PMID 32462740.
- ^ Office of the Commissioner (7 July 2017). «Press Announcements – FDA approves new treatment for sickle cell disease». www.fda.gov. Archived from the original on 10 July 2017. Retrieved 10 July 2017.
- ^ «Evidence-Based Management of Sickle Cell Disease» (PDF). 2014. Retrieved 16 November 2017.
twice-daily prophylactic penicillin beginning in early infancy and continuing through at least age 5
- ^ Dixit R, Nettem S, Madan SS, Soe HH, Abas AB, Vance LD, Stover PJ (March 2018). «Folate supplementation in people with sickle cell disease». The Cochrane Database of Systematic Reviews. 3 (4): CD011130. doi:10.1002/14651858.CD011130.pub3. PMC 5440187. PMID 29546732.
- ^ Oniyangi O, Omari AA (November 2019). «Malaria chemoprophylaxis in sickle cell disease». The Cochrane Database of Systematic Reviews. 2019 (11). doi:10.1002/14651858.CD003489.pub2. PMC 6532723. PMID 31681984.
- ^ Carroll CP (January 2020). «Opioid treatment for acute and chronic pain in patients with sickle cell disease». Neuroscience Letters. Elsevier BV. 714: 134534. doi:10.1016/j.neulet.2019.134534. PMID 31593753. S2CID 203667575.
- ^ Okomo U, Meremikwu MM (July 2017). «Fluid replacement therapy for acute episodes of pain in people with sickle cell disease». The Cochrane Database of Systematic Reviews. 7 (4): CD005406. doi:10.1002/14651858.CD005406.pub5. PMC 6483538. PMID 28759112.
- ^ Center for Drug Evaluation and Research (20 December 2019). «FDA approves crizanlizumab-tmca for sickle cell disease». FDA.
- ^ a b Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. (July 1998). «Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography». The New England Journal of Medicine. 339 (1): 5–11. doi:10.1056/NEJM199807023390102. PMID 9647873.
- ^ Kolata G (24 May 2021). «These Sisters With Sickle Cell Had Devastating, and Preventable, Strokes». New York Times.
- ^ Kolata G (24 May 2021). «These Sisters With Sickle Cell Had Devastating, and Preventable, Strokes». New York Times.
- ^ Aldrich TK, Nagel RL (1998). «Pulmonary Complications of Sickle Cell Disease.». In Reynolds HY, Bone RC, Dantzker DR, George RB, Matthay RA (eds.). Pulmonary and Critical Care Medicine (6th ed.). St. Louis: Mosby. pp. 1–10. ISBN 978-0-8151-1371-3.
- ^ Martí-Carvajal AJ, Conterno LO, Knight-Madden JM (September 2019). «Antibiotics for treating acute chest syndrome in people with sickle cell disease». The Cochrane Database of Systematic Reviews. 9 (4): CD006110. doi:10.1002/14651858.CD006110.pub5. PMC 6749554. PMID 31531967.
- ^ Dolatkhah R, Dastgiri S (January 2020). «Blood transfusions for treating acute chest syndrome in people with sickle cell disease». The Cochrane Database of Systematic Reviews. 1 (1): CD007843. doi:10.1002/14651858.CD007843.pub4. PMC 6984655. PMID 31942751.
- ^ a b Rankine-Mullings AE, Nevitt SJ (September 2022). «Hydroxyurea (hydroxycarbamide) for sickle cell disease». The Cochrane Database of Systematic Reviews. 2022 (10): CD002202. doi:10.1002/14651858.CD002202.pub3. PMC 9435593. PMID 36047926.
- ^ Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. (May 1995). «Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia». The New England Journal of Medicine. 332 (20): 1317–1322. doi:10.1056/NEJM199505183322001. PMID 7715639.
- ^ Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. (April 2003). «Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment». JAMA. 289 (13): 1645–1651. doi:10.1001/jama.289.13.1645. PMID 12672732.
- ^ Platt OS (March 2008). «Hydroxyurea for the treatment of sickle cell anemia». The New England Journal of Medicine. 358 (13): 1362–1369. doi:10.1056/NEJMct0708272. PMID 18367739. S2CID 351061.
- ^ Center for Drug Evaluation and Research (25 November 2019). «FDA approves voxelotor for sickle cell disease». FDA. Retrieved 9 December 2019.
- ^ Drasar E, Igbineweka N, Vasavda N, Free M, Awogbade M, Allman M, et al. (March 2011). «Blood transfusion usage among adults with sickle cell disease – a single institution experience over ten years». British Journal of Haematology. 152 (6): 766–770. doi:10.1111/j.1365-2141.2010.08451.x. PMID 21275951. S2CID 44562296.
- ^ Gyang E, Yeom K, Hoppe C, Partap S, Jeng M (January 2011). «Effect of chronic red cell transfusion therapy on vasculopathies and silent infarcts in patients with sickle cell disease». American Journal of Hematology. 86 (1): 104–106. doi:10.1002/ajh.21901. PMID 21117059.
- ^ Mirre E, Brousse V, Berteloot L, Lambot-Juhan K, Verlhac S, Boulat C, et al. (March 2010). «Feasibility and efficacy of chronic transfusion for stroke prevention in children with sickle cell disease». European Journal of Haematology. 84 (3): 259–265. doi:10.1111/j.1600-0609.2009.01379.x. PMID 19912310. S2CID 24316310.
- ^ Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, et al. (August 1996). «Bone marrow transplantation for sickle cell disease». The New England Journal of Medicine. 335 (6): 369–376. doi:10.1056/NEJM199608083350601. PMID 8663884. S2CID 25256772.
- ^ Kaiser J (5 December 2020). «CRISPR and another genetic strategy fix cell defects in two common blood disorders». ScienceMag.org. Science. Retrieved 7 December 2020.
… teams report that two strategies for directly fixing malfunctioning blood cells have dramatically improved the health of a handful of people with these genetic diseases.
- ^ Kumar V, Abbas AK, Fausto N, Aster J (28 May 2009). Robbins and Cotran Pathologic Basis of Disease (Professional Edition: Expert Consult – Online (Robbins Pathology) ed.). Elsevier Health. pp. Kindle Locations 33530–33531.
- ^ Wierenga KJ, Hambleton IR, Lewis NA (March 2001). «Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study». Lancet. 357 (9257): 680–683. doi:10.1016/s0140-6736(00)04132-5. PMID 11247552. S2CID 37012133.
- ^ Costa FF, Conran N (2016). Sickle Cell Anemia: From Basic Science to Clinical Practice. Springer. p. 35. ISBN 9783319067131. Retrieved 8 May 2016.
- ^ Prabhakar H, Haywood C, Molokie R (May 2010). «Sickle cell disease in the United States: looking back and forward at 100 years of progress in management and survival». American Journal of Hematology. 85 (5): 346–353. doi:10.1002/ajh.21676. PMID 20425797.
- ^ Weatherall DJ, Clegg JB (2001). «Inherited haemoglobin disorders: an increasing global health problem». Bulletin of the World Health Organization. 79 (8): 704–712. PMC 2566499. PMID 11545326.
- ^ Roberts I, de Montalembert M (July 2007). «Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe». Haematologica. 92 (7): 865–871. doi:10.3324/haematol.11474. PMID 17606434.
- ^ Wellems TE, Hayton K, Fairhurst RM (September 2009). «The impact of malaria parasitism: from corpuscles to communities». The Journal of Clinical Investigation. 119 (9): 2496–2505. doi:10.1172/JCI38307. PMC 2735907. PMID 19729847.
- ^ National Library of Medicine. URL = https://ghr.nlm.nih.gov/condition/sickle-cell-disease#statistics Archived 15 May 2016 at the Wayback Machine
- ^ WHO. «Sickle-cell anaemia – Report by the Secretariat» (PDF). Archived from the original (PDF) on 4 January 2011. Retrieved 27 November 2010.
- ^ Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, et al. (April 2002). «Protective effects of the sickle cell gene against malaria morbidity and mortality». Lancet. 359 (9314): 1311–1312. doi:10.1016/S0140-6736(02)08273-9. PMID 11965279. S2CID 37952036.
- ^ Tusuubira SK, Nakayinga R, Mwambi B, Odda J, Kiconco S, Komuhangi A (April 2018). «Knowledge, perception and practices towards sickle cell disease: a community survey among adults in Lubaga division, Kampala Uganda». BMC Public Health. 18 (1): 561. doi:10.1186/s12889-018-5496-4. PMC 5924488. PMID 29703184.
- ^ Ndeezi G, Kiyaga C, Hernandez AG, Munube D, Howard TA, Ssewanyana I, et al. (March 2016). «Burden of sickle cell trait and disease in the Uganda Sickle Surveillance Study (US3): a cross-sectional study». The Lancet. Global Health. 4 (3): e195–e200. doi:10.1016/S2214-109X(15)00288-0. PMID 26833239.
- ^ National Heart, Lung and Blood Institute. «Sickle cell anemia, key points». Archived from the original on 2 December 2010. Retrieved 27 November 2010.
- ^ «Data & Statistics on Sickle Cell Disease | CDC». Centers for Disease Control and Prevention. 31 August 2016. Retrieved 13 December 2019.
- ^ «September is Sickle Cell Awareness Month». CDC. Archived from the original on 27 September 2010. Retrieved 6 February 2011.
- ^ «Sickle Cell Trait». www.hematology.org. 8 September 2017. Retrieved 13 December 2019.
- ^ «Disorder Name: Sickle Cell Disease». New Born Screening. Archived from the original on 28 September 2016. Retrieved 11 October 2016.
- ^ «default – Stanford Children’s Health». www.stanfordchildrens.org. Retrieved 14 March 2020.
- ^ Edwards QT, Seibert D, Macri C, Covington C, Tilghman J (November 2004). «Assessing ethnicity in preconception counseling: genetics—what nurse practitioners need to know». Journal of the American Academy of Nurse Practitioners. 16 (11): 472–480. doi:10.1111/j.1745-7599.2004.tb00426.x. PMID 15617360. S2CID 7644129.
- ^ «Sickle Cell Patients Endure Discrimination, Poor Care And Shortened Lives». NPR.org. 4 November 2017. Retrieved 12 November 2017.
- ^ Bardakdjian J, Wajcman H (September 2004). «[Epidemiology of sickle cell anemia]». La Revue du Praticien (in French). 54 (14): 1531–1533. PMID 15558961.
- ^ Thuret I, Sarles J, Merono F, Suzineau E, Collomb J, Lena-Russo D, et al. (June 2010). «Neonatal screening for sickle cell disease in France: evaluation of the selective process». Journal of Clinical Pathology. 63 (6): 548–551. doi:10.1136/jcp.2009.068874. PMID 20498028. S2CID 22391674.
- ^ «Inheriting sickle cell anaemia – Live Well – NHS Choices». www.nhs.uk. 23 October 2017. Archived from the original on 2 December 2014.
- ^ «Sickle cell anaemia – NHS Choices». www.nhs.uk. 23 October 2017. Archived from the original on 13 December 2011.
- ^ «Who is offered screening and when?». screening.nhs.uk. Archived from the original on 31 December 2014.
- ^ «Give Blood – Resources – Sickle Cell and Blood Donation». Give Blood. Archived from the original on 31 December 2014.
- ^ «Why is Blood from Afro-Caribbean Donors Special?». sicklecellsociety.org. Archived from the original on 30 December 2014.
- ^ Jastaniah W (2011). «Epidemiology of sickle cell disease in Saudi Arabia». Annals of Saudi Medicine. 31 (3): 289–293. doi:10.4103/0256-4947.81540. PMC 3119971. PMID 21623060.
- ^ Memish ZA, Saeedi MY (2011). «Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and β-thalassemia in Saudi Arabia». Annals of Saudi Medicine. 31 (3): 229–235. doi:10.4103/0256-4947.81527. PMC 3119961. PMID 21623050.
- ^ Al Arrayed S (1995). «Features of sickle-cell disease in Bahrain». Eastern Mediterranean Health Journal. 1 (1). Archived from the original on 8 October 2016.
- ^ Al Arrayed S, Al Hajeri A (2010). «Public awareness of sickle cell disease in Bahrain». Annals of Saudi Medicine. 30 (4): 284–288. doi:10.4103/0256-4947.65256. PMC 2931779. PMID 20622345.
- ^ «Sickle Cell Anemia». www.hematology.org. 16 December 2014. Archived from the original on 25 June 2017. Retrieved 1 May 2017.
- ^ Awasthy N, Aggarwal KC, Goyal PC, Prasad MS, Saluja S, Sharma M (2008). «Sickle cell disease: Experience of a tertiary care center in a nonendemic area». Annals of Tropical Medicine and Public Health. 1 (1): 1–4. doi:10.4103/1755-6783.43069.
- ^ «Life with sickle cell – Nation – Nepali Times». Archived from the original on 24 June 2015.
- ^ Asnani MR, McCaw-Binns AM, Reid ME (2011). «Excess risk of maternal death from sickle cell disease in Jamaica: 1998–2007». PLOS ONE. 6 (10): e26281. Bibcode:2011PLoSO…626281A. doi:10.1371/journal.pone.0026281. PMC 3200316. PMID 22039456.
- ^ Lebby R (1846). «Case of absence of the spleen». Southern J of Med Pharmacol. 1: 481–3.
- ^ a b c Ballas SK, Gupta K, Adams-Graves P (November 2012). «Sickle cell pain: a critical reappraisal». Blood. 120 (18): 3647–3656. doi:10.1182/blood-2012-04-383430. PMID 22923496.
- ^ Herrick JB (1 November 1910). «Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia». Archives of Internal Medicine. 6 (5): 179–184. doi:10.1001/archinte.1910.00050330050003.; reprinted as Herrick JB (2001). «Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910». The Yale Journal of Biology and Medicine. 74 (3): 179–184. PMC 2588723. PMID 11501714.
- ^ Washburn RE (1911). «Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia». The Virginia Medical Semi-Monthly. 15 (21): 490–493.
- ^ «UVa Hospital Celebrating 100 Years». University of Virginia. Archived from the original on 31 January 2015. Retrieved 28 January 2015.
- ^ Mason VR (1922). «Sickle cell anemia». JAMA. 79 (16): 1318–1320. doi:10.1001/jama.1922.02640160038012. Reprinted in Mason VR (October 1985). «Landmark article Oct. 14, 1922: Sickle cell anemia. By V.R. Mason». JAMA. 254 (14): 1955–1957. doi:10.1001/jama.254.14.1955. PMID 3900438.
- ^ Pauling L, Itano HA (November 1949). «Sickle cell anemia a molecular disease». Science. 110 (2865): 543–548. Bibcode:1949Sci…110..543P. doi:10.1126/science.110.2865.543. PMID 15395398. S2CID 31674765.
- ^ Ingram VM (October 1956). «A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin». Nature. 178 (4537): 792–794. Bibcode:1956Natur.178..792I. doi:10.1038/178792a0. PMID 13369537. S2CID 4167855.
- ^ «Foster, Gloria». Facts On File History Database. Archived from the original on 5 March 2016. Retrieved 25 February 2015.
- ^ Richard-Lenoble D, Toublanc JE, Zinsou RD, Kombila M, Carme B (1980). «[Results of a systematic study of drepanocytosis in 1,500 Gabonese using hemoglobin electrophoresis]» [Results of a systematic study of drepanocytosis in 1,500 Gabonese using hemoglobin electrophoresis]. Bulletin de la Société de Pathologie Exotique et de ses Filiales (in French). 73 (2): 200–206. PMID 7460122.
- ^ «Social Security Ruling: SSR 2017-3p». Office of Disability Policy. U.S. Social Security Administration. Retrieved 15 January 2018.
- ^ «Federal Register, Volume 82 Issue 178 (Friday, September 15, 2017)». www.gpo.gov. Retrieved 15 January 2018.
- ^ «Sickle Cell Disease | National Heart, Lung, and Blood Institute (NHLBI)». www.nhlbi.nih.gov. Retrieved 4 July 2020.
- ^ Bulgin D, Tanabe P, Jenerette C (August 2018). «Stigma of Sickle Cell Disease: A Systematic Review». Issues in Mental Health Nursing. 39 (8): 675–686. doi:10.1080/01612840.2018.1443530. PMC 6186193. PMID 29652215.
- ^ Washington HA (2006). Medical apartheid : the dark history of medical experimentation on Black Americans from colonial times to the present (1st paperback ed.). New York: Harlem Moon. ISBN 978-0-7679-1547-2. OCLC 192050177.
- ^ a b c d e f g h Tusuubira SK, Naggawa T, Nakamoga V (October 2019). «To Join Or Not To Join? A Case Of Sickle Cell Clubs, Stigma And Discrimination In Secondary Schools In Butambala District, Uganda». Adolescent Health, Medicine and Therapeutics. 10: 145–152. doi:10.2147/AHMT.S223956. PMC 6778728. PMID 31632168.
- ^ a b Tusuubira SK, Nakayinga R, Mwambi B, Odda J, Kiconco S, Komuhangi A (April 2018). «Knowledge, perception and practices towards sickle cell disease: a community survey among adults in Lubaga division, Kampala Uganda». BMC Public Health. 18 (1): 561. doi:10.1186/s12889-018-5496-4. PMC 5924488. PMID 29703184.
- ^ a b c d e f g «Sickle Cell Association of Uganda». Sickle Cell Association of Uganda. Retrieved 7 April 2021.
- ^ Okwi AL, Byarugaba W, Ndugwa CM, Parkes A, Ocaido M, Tumwine JK (September 2009). «Knowledge gaps, attitude and beliefs of the communities about sickle cell disease in Eastern and Western Uganda». East African Medical Journal. 86 (9): 442–449. doi:10.4314/eamj.v86i9.54167. PMID 21644415.
- ^ a b c d e Marsh VM, Kamuya DM, Molyneux SS (August 2011). «‘All her children are born that way’: gendered experiences of stigma in families affected by sickle cell disorder in rural Kenya». Ethnicity & Health. 16 (4–5): 343–359. doi:10.1080/13557858.2010.541903. PMC 3534410. PMID 21797722.
- ^ «Our Vision and Mission – Uganda Sickle Cell Rescue Foundation». 13 May 2017. Retrieved 6 April 2021.
- ^ a b «Background – Uganda Sickle Cell Rescue Foundation». 13 May 2017. Retrieved 6 April 2021.
- ^ Sehlo MG, Kamfar HZ (April 2015). «Depression and quality of life in children with sickle cell disease: the effect of social support». BMC Psychiatry. 15 (1): 78. doi:10.1186/s12888-015-0461-6. PMC 4394397. PMID 25880537.
- ^ «London Eye: Shouldn’t need an improvement programme». Health Service Journal. 27 July 2022. Retrieved 28 September 2022.
- ^ «Better protection and support for people with sickle cell disease». The Voice. October 2022. pp. 16–17. Retrieved 7 October 2022.
- ^ Rylatt, Aidan (7 October 2022). «No-one is Listening» (PDF).
- ^ «NHS launches lifesaving sickle cell campaign». 7 October 2022.
- ^ a b Kassim AA, Sharma D (December 2017). «Hematopoietic stem cell transplantation for sickle cell disease: The changing landscape». Hematology/Oncology and Stem Cell Therapy. 10 (4): 259–266. doi:10.1016/j.hemonc.2017.05.008. PMID 28641096.
- ^ a b Olowoyeye A, Okwundu CI (November 2020). «Gene therapy for sickle cell disease». The Cochrane Database of Systematic Reviews. 2020 (11): CD007652. doi:10.1002/14651858.CD007652.pub7. PMC 8275984. PMID 33251574.
- ^ Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, et al. (December 2001). «Correction of sickle cell disease in transgenic mouse models by gene therapy». Science. 294 (5550): 2368–2371. Bibcode:2001Sci…294.2368P. doi:10.1126/science.1065806. PMID 11743206. S2CID 25607771.
- ^ Wilson JF (18 March 2002). «Murine Gene Therapy Corrects Symptoms of Sickle Cell Disease». The Scientist – Magazine of the Life Sciences. Retrieved 17 December 2014.
- ^ St. Jude Children’s Research Hospital (4 December 2008). «Gene Therapy Corrects Sickle Cell Disease In Laboratory Study». ScienceDaily. Archived from the original on 13 December 2014. Retrieved 17 December 2014.
- ^ Clinical trial number NCT02247843 for «Stem Cell Gene Therapy for Sickle Cell Disease» at ClinicalTrials.gov
- ^ Clinical trial number NCT00012545 for «Collection and Storage of Umbilical Cord Stem Cells for Treatment of Sickle Cell Disease» at ClinicalTrials.gov
- ^ Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. (March 2017). «Gene Therapy in a Patient with Sickle Cell Disease». The New England Journal of Medicine. 376 (9): 848–855. doi:10.1056/NEJMoa1609677. PMID 28249145. S2CID 5128871.
- ^ Kolata G (27 January 2019). «These Patients Had Sickle-Cell Disease. Experimental Therapies Might Have Cured Them». The New York Times. Retrieved 28 January 2019.
- ^ Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, et al. (November 2016). «CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells». Nature. 539 (7629): 384–389. Bibcode:2016Natur.539..384D. doi:10.1038/nature20134. PMC 5898607. PMID 27820943.
- ^ «In A 1st, Doctors In U.S. Use CRISPR Tool To Treat Patient With Genetic Disorder». NPR.org. Retrieved 31 July 2019.
- ^ Zipkin, Mark (6 December 2019). «CRISPR’s «magnificent moment» in the clinic». Nature Biotechnology. doi:10.1038/d41587-019-00035-2.
- ^ Persaud A, Desine S, Blizinsky K, Bonham VL (August 2019). «A CRISPR focus on attitudes and beliefs toward somatic genome editing from stakeholders within the sickle cell disease community». Genetics in Medicine. 21 (8): 1726–1734. doi:10.1038/s41436-018-0409-6. PMC 6606394. PMID 30581191.
- ^ Walker M (15 January 2018). «Gene Therapy». Sickle Cell Disease News. Retrieved 14 March 2020.
- ^ Oringanje C, Nemecek E, Oniyangi O (July 2020). «Hematopoietic stem cell transplantation for people with sickle cell disease». The Cochrane Database of Systematic Reviews. 2020 (7): CD007001. doi:10.1002/14651858.CD007001.pub5. PMC 7390490. PMID 32617981.
Further reading[edit]
- Brown RT, ed. (2006). Comprehensive handbook of childhood cancer and sickle cell disease: a biopsychosocial approach. Oxford University Press. ISBN 978-0-19-516985-0.
- Hill SA (2003). Managing Sickle Cell Disease in Low-Income Families. Temple University Press. ISBN 978-1-59213-195-2.
- Serjeant GR, Serjeant BE (2001). Sickle Cell Disease. Oxford University Press. ISBN 978-0-19-263036-0.
- Tapper M (1999). In the blood: sickle cell anemia and the politics of race. University of Pennsylvania Press. ISBN 978-0-8122-3471-8.
External links[edit]
- Sickle cell at Curlie
Серповидноклеточная анемия
Лечением данного заболевания занимается Гематолог
Информация, представленная на странице, не должна быть использована для самолечения или самодиагностики. При подозрении на наличие заболевания, необходимо обратиться за помощью к квалифицированному специалисту. Провести диагностику и назначить лечение может только ваш лечащий врач.
Содержание статьи:
- Причины
- Виды
- Симптомы
- Осложнения
- Диагностика
- Лечение
- Прогноз и профилактика
Серповидно-клеточной анемией называют наследственную гемоглобинопатию, которая проявляется выработкой аномального гемоглобина S, изменением свойств и формы эритроцитов Источник:
Серповидноклеточная анемия. Доклад секретариата. Пятьдесят девятая сессия a59/9 всемирной ассамблеи здравоохранения. 2016. с.1-7.
Симптомами серповидно-клеточной анемии являются апластические и гемолитические кризы, закупорка сосудов тромбами, боли в суставах и костях, увеличение селезенки.
Диагноз ставится на основе результатов исследования крови пациента и пункции костного мозга. Лечение симптоматическое, консервативное, направлено на снятие и профилактику кризов. Пациенту назначают прием антикоагулянтов, проводят переливание эритроцитов. При наличии показаний проводится операция по удалению селезенки.
Причины
Данный вид заболевания возникает из-за генетических аномалий, которые приводят к синтезу серповидного гемоглобина. Это патологическая форма. Когда из такого гемоглобина высвобождается кислород он склеивается, что приводит к образованию стержней, которые меняют форму эритроцита и повреждают его. Именно такие патологические изменения приводят к развитию симптоматики заболевания.
Данная патология является наследственной и не заразна для окружающих людей. Заболевание возникает, если малыш наследует аномальные гены от обоих родителей. Если унаследован аномальный ген от одного родителя, то человек является носителем, но сам не болеет. Анемии у него не возникает.
Виды
Выделяют два вида данной патологии:
1. Гомозиготная. В гомозиготах имеется по одному аномальному гену от отца и матери. В крови таких пациентов только серповидные эритроциты, в связи с чем болезнь развивается быстро и отличается тяжелым течением.
2. Гетерозиготная. Патологический ген наследуется только от одного из родителей. Поэтому в крови имеются не только аномальные, но и нормальные эритроциты. У таких пациентов клинические признаки заболевания возникают редко, при наличии определенных условий.
Симптомы
Клиника серповидной анемии обычно проявляется в раннем детстве (в течение первых двух лет). Основными симптомами являются бледность кожи и слизистых оболочек, увеличение селезенки, задержка развития, слабый иммунитет что приводит к частым инфекционным заболеваниям Источник:
К вопросу о клинических проявлениях серповидноклеточного заболевания. Кузьмин Г.Д., Троицкая О.В., Мансур Т.И., Кузнецов В.И., Маслова А.Н. Вестник РУДН. Серия: Медицина. 2005. №1. с.81-84. Помимо этого, наблюдается болезненные припухлости стоп и кистей, что является следствием закупорки капилляров в мелких костях.
Вследствие тромбозов происходит закупорка тканей по всему организму. Это может привести к развитию инсульта, некроза почек и костей, инфаркта селезенки, образованию язв на нижних конечностях, снижению зрения, болезненной патологической эрекции у мужчин.
Закупорка сосудов костей приводит к появлению сильных болей. Нарушение функции селезенки увеличивает частоту заболеваемости бактериальными инфекциями и может привести к развитию тяжелых осложнений (остеомиелит, сепсис и другие).
Основной причиной смерти пациентов всех возрастных групп является инфекция. У людей среднего возраста летальный исход может наступить на фоне дыхательной и почечной недостаточности.
Гетерозиготные пациенты, которые унаследовали дефектный ген только у одного родителя обычно являются клинически здоровыми. Но если возникает серьезная гипоксия (например, при подъеме в гору) некоторые эритроциты могут принимать форму серпа, что приводит к появлению симптомов данного вида заболевания.
Хроническое течение серповидно-клеточной формы с частыми кризами приводит к развитию в организме необратимых изменений, которые могут стать причиной гибели пациента. У некоторых больных отмечается сморщивание селезенки, вызванное аутоиммунными факторами. На этом фоне сильно снижается иммунитет, у пациентов часто возникают тяжело протекающие инфекции Источник:
Серповидно-клеточная анемия: случай из клинической практики. Шапченко А.В., Арабидзе Г.Г., Муслимова О.В., Фищенко С.Г., Скрябина Е.О., Полякова О.В. Нескаромная С.А. CardioСоматика. 2013. №3. с.52-58 (менингит, пневмония, сепсис и ряд других).
Закупорка кровеносных сосудов приводит к развитию кризов. В итоге даже у детей могут возникать ишемические инсульты. У взрослых людей чаще наблюдаются кровоизлияния в мозг, эректильная дисфункция, поражение сосудов сетчатки, почечная недостаточность.
У женщин с данной формой заболевания позднее начинаются месячные, имеется предрасположенность к выкидышам и преждевременным родам. Может развиться сердечная и хроническая почечная недостаточность.
В результате длительного разрушения эритроцитов с выбросом в кровь гемоглобина и образования излишков билирубина развивается желчнокаменная болезнь, холецистит. Помимо этого, у пациентов с данной формой патологии могут возникать такие осложнения, как остеомиелит, некроз костей, язвы голеней.
Диагностика
Диагноз ставится после лабораторного изучения свойств гемоглобина пациента. Самой старой методикой выявления болезни является исследование «влажного мазка». Суть процедуры заключается в том, что, если смочить мазок крови специальным веществом эритроциты начинают быстро отдавать кислород и принимают характерную патологическую форму. Чтобы результат был максимально достоверным, через сутки делается повторный анализ.
Еще один, более распространенный метод, основан на выявлении патологических элементов крови по их сниженной растворимости в некоторых растворах, по сравнению с нормальными клетками. Большая популярность данной методики связана с тем, что результаты исследования можно получить в течение 15 минут.
Важно! Вышеприведенные исследования позволяют определить наличие патологии, но не выявляют ее форму (гомозиготная или гетерозиготная), что крайне важно знать в плане дальнейшего прогноза. Выявить форму болезни на сегодняшний день можно только при помощи анализа подвижности гемоглобина в электрическом поле. Без данного исследования точная диагностика невозможна. Но для массовых обследований данная методика не подходит, так как занимает большое количество времени и дорого стоит.
Для терапии данной формы заболевания используются:
- сильные наркотические обезболивающие (морфин и другие);
- антибактериальные препараты (для лечения инфекционных осложнений);
- НПВС (для снятия воспаления);
- кислородотерапия;
- введение специальных растворов (через рот или внутривенно).
При тяжелом течении болезни или перед оперативным вмешательством проводится переливание эритроцитов. В ряде случаев для проведения данной манипуляции используется специальный аппарат, который удаляет патологические измененные клетки и заменяет их на здоровые.
Для снижения тяжести и частоты болевых приступов, а также сокращения количества госпитализаций и переливаний крови пациенту может быть назначен прием цитостатиков Источник:
Серповидно-клеточное заболевание и его неврологические проявления. Кузьмин Г.Д., Троицкая О.В., Мансур Т.И., Юшкова Н.М., Кузнецов В.И. Вестник РУДН. Серия: Медицина. 2004. №3. с.34-36.
Полностью вылечить заболевание можно только при помощи пересадки пациенту стволовых клеток. Это относительно новая методика. Ее проведение сопряжено с серьезными рисками (у некоторых пациентов возможен летальный исход). Но если пересадка проходит успешно, то человек полностью выздоравливает. Трансплантацию обычно делают молодым пациентам с тяжелой формой заболевания, которым удалось найти донора.
Сейчас клинические испытания проходит еще ряд эффективных методик лечения данной патологии. Поэтому есть высокая вероятность, что через несколько лет на фармацевтическом рынке появятся новые эффективные препараты для терапии серповидно-клеточной формы болезни.
Прогноз и профилактика
На сегодняшний день заболевание с трудом поддается лечению. У детей старше 10 лет количество кризов начинает снижаться, но развиваются серьезные осложнения. Ряд пациентов умирают в детском возрасте из-за сепсиса или некроза тканей. Большинство больных доживают в среднем до 50 лет Источник:
Анемии. Сараева Н.О. Учебное пособие для студентов. Иркутский государственный медицинский университет Федерального агентства по здравоохранению и социальному развитию. 2009. с.78-80.
Специфической профилактики нет, так как патология имеет наследственный характер. Чтобы замедлить прогрессирование заболевания, нужно стараться избегать инфекций, обезвоживания организма, хронического стресса, нервного перенапряжения, кислородного голодания, воздействия слишком высоких или низких температур.
Детям с данной формой гемолитической анемии показана обязательная вакцинация против менингококка и пневмококка. Если в семье имеются люди, больные данной формой заболевания, то при планировании беременности необходима консультация генетика для оценки возможных рисков для будущего ребенка.
- К вопросу о клинических проявлениях серповидноклеточного заболевания. Кузьмин Г.Д., Троицкая О.В., Мансур Т.И., Кузнецов В.И., Маслова А.Н. Вестник РУДН. Серия: Медицина. 2005. №1. с.81-84
- Серповидно-клеточная анемия: случай из клинической практики. Шапченко А.В., Арабидзе Г.Г., Муслимова О.В., Фищенко С.Г., Скрябина Е.О., Полякова О.В. Нескаромная С.А. CardioСоматика. 2013. №3. с.52-58
- Серповидно-клеточное заболевание и его неврологические проявления. Кузьмин Г.Д., Троицкая О.В., Мансур Т.И., Юшкова Н.М., Кузнецов В.И. Вестник РУДН. Серия: Медицина. 2004. №3. с.34-36
- Анемии. Сараева Н.О. Учебное пособие для студентов. Иркутский государственный медицинский университет Федерального агентства по здравоохранению и социальному развитию. 2009. с.78-80
- Серповидноклеточная анемия. Доклад секретариата. Пятьдесят девятая сессия a59/9 всемирной ассамблеи здравоохранения. 2016. с.1-7
Статья опубликована: 31.01.2023 г.
Последнее обновление: 21.02.2023 г.
Серповидно-клеточные нарушения
![]()
Версия: Клинические рекомендации РФ 2021 (Россия)
Категории МКБ:
Бета-талассемия (D56.1), Серповидно-клеточные нарушения (D57)
Разделы медицины:
Гематология, Гематология детская
Общая информация
Краткое описание
Разработчик клинической рекомендации:
Национальное гематологическое общество
Национальное общество детских гематологов и онкологов
Одобрено на заседании научно-практического совета Министерства здравоохранения Российской Федерации (протокол от «31» августа 2021 г. № 18/2-3-4)
Клинические рекомендации
Серповидно-клеточные нарушения
Возрастная группа: взрослые/дети
Год утверждения: 2021 г.
Определение заболевания или состояния (группы заболеваний или состояний)
Серповидно-клеточная болезнь (СКБ) – группа состояний, обусловленных наличием специфических мутаций в бета-глобиновом гене, приводящих к изменению физико-химических свойств гемоглобина (полимеризация в деокси-форме, нарушение сродства гемоглобина к кислороду). К СКБ относится серповидно-клеточная анемия (гомозиготность по аномальному гемоглобину S (HbSS; результат замены в 6 позиции бета-глобина валина на глютамин) и другие состояния, развившиеся вследствие сонаследования аномального HbS с другими поломками бета-глобинового гена, приводящими к количественному уменьшению нормального бета-глобина (бета-талассемия), (S-β-талассемия) или другому качественному нарушению бета-глобина [1–8].
Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
D57 – серповидно-клеточные нарушения
D57.0 – серповидно-клеточная анемия с кризом (HbSS болезнь с кризом)
D57.1 – серповидно-клеточная анемия без криза
D57.2 – двойные гетерозиготные серповидно-клеточные нарушения (болезнь HbSC, HbSD, HbSE и др.)
D57.3 – Носительство признака серповидно-клеточности
D57.8 – другие серповидно-клеточные нарушения
D56.1 – серповидно-клеточная бета-талассемия
Облачная МИС «МедЭлемент»
Облачная МИС «МедЭлемент»

Автоматизация клиники: быстро и недорого!
- Подключено 500 клиник из 4 стран
- 1 место — 800 RUB / 5500 KZT / 27 BYN в месяц
Классификация
Классификация заболевания или состояния (группы заболеваний или состояний)
В настоящее время СКБ подразделяется в зависимости от наличия, или отсутствия криза и специфического генотипа [1–6].
- серповидно-клеточная анемия с кризом;
- серповидно-клеточная анемия без криза;
- серповидно-клеточные нарушения с кризом;
- серповидно-клеточные нарушения без криза;
- серповидно-клеточная бета-талассемия с кризом;
- серповидно-клеточная бета-талассемия без криза.
Этиология и патогенез
Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
Аномальный HbS полимеризуется в деоксиформе, теряя растворимость. Полимеризация гемоглобина зависит от концентрации HbS внутри эритроцитов, степени деоксигенации гемоглоибна, рН и внутриклеточной концентрации HbF. Полимеры HbS взаимодействуют с эритроцитарной мембраной, меняя внешний вид эритроцитов на серповидные. Серповидные эритроциты окклюзируют микроциркуляроное русло, приводя к повреждению сосуда, инфарктам органов, болевым кризам и другим симптомам, ассоциированным с СКБ [3–9].
При СКБ одновременно протекает два патологических процесса: гемолиз и вазо-окклюзия [3–9]:
— Гемолиз приводит к анемии и функциональному дефициту оксида азота, что в свою очередь приводит к повреждению эндотелия сосудов, развитию пролиферативной эндотелиопатии и таких осложнений как легочная гипертензия, приопизм и инсульт.
— Вазо-окклюзия вызывает острую и хроническую ишемию тканей, что проявляется острой болью (болевой криз) и вызывает повреждение органов.
Эпидемиология
Эпидемиология заболевания или состояния (группы заболеваний или состояний)
По данным ВОЗ в мире болеют СКБ >4 миллионов человек, ежегодно только в Африканских странах рождается 6-9 миллионов пациентов СКБ [10]. Около 80% пациентов СКБ – африканцы, достаточно часто СКБ встречается в Индии и арабских странах, а также среди выходцев из этих стран, проживающих в других регионах (страны северной Америки, европейские страны, Великобритания), так в США СКБ встречается с частотой 1 случай на 5000 населения, во Франции – 1 случай на 2500 населения, Великобритании – 1 случай на 2000 населения [10]. Для Российской Федерации СКБ – редкое заболевание, немногочисленные случаи встречаются в основном в пограничных с республикой Азербайджан областях [6]. В среднем пациенты СКБ в развитых странах доживают до 30-40 лет [10].
Клиническая картина
Cимптомы, течение
Клиническая картина заболевания или состояния (группы заболеваний или состояний)
Бледность кожи и слизистых оболочек, иногда легкая иктеричность, появляются в возрасте 3-6 мес. и усиливаются с возрастом.
Спленомегалия может присутствовать у некоторых пациентов в возрасте старше 6 месяцев, но у большинства к 6-8 годам наступает аспления вследствие вазо-окклюзивных кризов.
Гепатомегалия может присутствовать у пациентов первых 10 лет жизни.
Болевой синдром различной локализации и интенсивности у пациентов старше 1 года вследствие вазо-окклюзивных кризов.
Субфибрилитет / фебрильная лихорадка вследствие вазо-окклюзивных кризов у пациентов старше 1 года.
Отставание в физическом и половом развитии в детском и подростковом возрасте.
Развитие тех или иных осложнений СКБ сопровождается расширением клинических проявлений.
Основная причина смертности и инвалидизации пациентов СКБ, особенно в детском возрасте – инсульты, которые встречаются у 5-10% детей с СКБ, «немые» инсульты встречаются до 20% пациентов СКБ в возрасте до 20%. Второй существенной причиной инвалидизации и смертности при СКБ является острый грудной синдром (ОГС), который развивается вследствие инфекции, инфаркта легкого или их комбинации, и острая секвестрация в селезенке [11–21]. Другие причины инвалидизации – поражения легких, печени, почек и глаз, приапизм, аваскулярный некроз, остеомиелит, трофические язвы нижних конечностей [11–21]. Одной из основных причин госпитализации при СКБ являются болевые кризы любой локализации и интенсивности [1–8]. Жизнеугрожающие остро возникающие осложнения СКБ – фульминантный сепсис, острый грудной синдром (ОГС), острая секвестрация в селезенке или печени, ишемический или геморрагический инсульт, субарахноидальное кровоизлияние, ОПН, СОПОН, синдром билиарной обструкции (внутрипеченочный холестаз), фульминантный приапизм, посттрансфузионный синдром гипергемолиза, острые офтальмологичесике осложнения, остеонекроз крупный суставов (например, тазобедренных, плечевых) [1,2,11–21].
Вследствие многократных вазо-окклюзивных кризов селезенки развивается аспления, что сопровождается резким возрастанием риска смерти от инфекций, особенно пневмококковой [1–6,22]. Инсульт – одно из основных осложнений СКБ, сопровождающее значительной инвалидизацией пациентов. У пациентов, перенесших инсульт, страдает когнитивная функция и нарушается социальная адаптация. Кооперативные исследования СКБ показали преимущественное развитие инсульта у пациентов с HbSS (в 4 раза выше, чем у пациентов с HbSC и др.), причем преимущественно в возрасте 2-5 лет, заболеваемость инсультами в США среди пациентов с HbSS составляет 0,6 на 100 пациенто-лет, к 20 годам – 11% всех пациентов СКБ [1–6,8,12,18–21,23].
У пациентов СКБ очень часто отмечаются различные отклонения от нормы сердечно-сосудистой системы не зависимо от возраста: кардиомегалия и гиперактивный прекардиальной отдел, систолические шумы (у большинства пациентов), преждевременные сокращения (часто присутствуют у взрослых пациентов). Физическая работоспособность снижается примерно до половины у взрослых пациентов с СКБ и от 60 до 70 процентов у детей, что связано с тяжестью анемии [1–6,24,25]. Кардиомегалия и шумы в сердце часто поднимают вопрос о наличии застойной сердечной недостаточности. Сократимость миокарда нормальная, и явная застойная сердечная недостаточность бывает редко, особенно у детей. Когда сердечная недостаточность присутствует, она часто связана с вторичными причинами, такими как перегрузка жидкостью. Сердечный выброс увеличивается в покое и в дальнейшем возрастает с физической нагрузкой [24–26]. ЭКГ часто имеют неспецифические нарушения, например, признаки увеличение желудочков. В состоянии покоя сердечный индекса в 1,5 раза выше нормального значения, и увеличивается больше во время физической нагрузки, что не полностью объясняется содержанием Hb или кислорода, указывая тем самым на повышение потребления кислорода миокардом [1–6,8,12,14,24–26].
Пациенты старшего детского возраста часто имеют увеличение размера полости левого и правого желудочков и левого предсердия, увеличение толщины межжелудочковой перегородки и нормальную сократимость миокарда. Эти изменения, за исключением правого желудочка, были обратно пропорциональны Hb и указывают на дилатацию левых отделов сердца, что также зависит от возраста. При наличии гомозиготной α-талассемии-2, размеры левого желудочка были более нормальные, но толщина стенок была увеличена. Это различие было является результатом более высокого Hb вследствие α-талассемии. Тем не менее, улучшения толерантности к физической нагрузке не было отмечено, возможно, из-за аномальных свойств серповидных эритроцитов [24–26]. Выпот в перикарде присутствовал у 10% пациентов, что также находится в обратной зависимости от Hb [24].
Артериальное давление у пациентов с СКБ выше, чем ожидается при имеющейся у них степени тяжести анемии, что предполагает наличие относительной артериальной гипертензии [27,28]. Небольшое исследование показало взаимосвязь между повышением АД и развитием инсульта. Выживаемость снижается, риск развития инсульта растет при повышении АД. Что позволило предположить патогенетическую важность «относительной» артериальной гипертензии. Увеличение продолжительности жизни, более высокой распространенности серповидно-клеточной нефропатии, потребление высококалорийной, высоко-солевой диеты, все это, вероятно, способствует увеличению встречаемости абсолютной гипертензии у пациентов с СКБ. Если это прибавить к очевидным рискам даже относительной артериальной гипертензии и факту, что снижение артериального давления у людей без СКА может предотвратить последствие гипертонии, представляется разумным рассмотреть антигипертензивную терапию у пациентов с СКБ с пограничной гипертензией [27,28].
Инфаркт миокарда у пациентов СКБ встречается достаточно редко, окклюзия коронарных артерий также крайне редка [29], что позволяет предположить, что васкулопатия мелких сосудов приводит к поражению сердца. У пациентов с СКБ может быть ишемия миокарда, поэтому при жалобах на боль в груди необходимо ее исключать [29,30].
У взрослых пациентов с СКБ часто бывает внезапная смерть, что связано с дисфункцией проводящей системы сердца [30].
ОГС – остро возникшее заболевание, характеризующееся лихорадкой (выше 38,50С) и респираторынми симптомами (укорочение дыхания, тахипноэ, шумное дыхание, хрипы, ослабление дыхания), сопровождающимися свежими инфильтративными изменениями в легких при рентгенографии [1–6,31,32]. Поскольку появление радиологических изменений может отставать, диагноз устанавливается не сразу. Основной фактор риска развития ОГС – генотип СКБ: наиболее часто встречается при HbSS, наиболее редко при S/β+-талассемии [31,33,34]. ОГС – наиболее частая причина хирургических вмешательств и анестезии у пациентов СКБ [33]. Дети имеют более высокую частоту развития ОГС, но при этом более низкую смертность (<2%), чем взрослые (4-9%) [31,33]. У пациентов HbSS случаи развития ОГС ассоциированы с низким HbF, высоким гематокритом, лейкоцитозом и отсутствием сонаследования α-талассемии. У детей отмечается сезонность в частоте развития ОГС: ниже летом, чаще зимой, когда высокая вероятность острых респираторных заболеваний. У взрослых пациентов СКБ сезонность развития ОГС почти отсутствует. ОГС может протекать с или без прогрессирующей дыхательной недостаточности, характеризующейся некардиологическим отеком легких и тяжелой гипоксемией [31–34].
В большом клиническом исследовании [33,35], среди причин, вызвавших развитие ОГС, были отмечены жировая эмболия легких (16,2% случаев развития ОГС; у пожилых пациентов с низкой сатурацией кислорода – 44-77% случаев развития ОГС), инфекции (54% случаев развития ОГС). Спектр инфекционных агентов – хламидии (13%), микоплазма (12%), различные вирусы (12%), бактерии, включая St. aureus, Str. pneumonia, Hemophilia influenza (8,2%).
Кратковременный прогноз ОГС с ограниченным вовлечением легких и легкой гипоксемией хороший. Есть сообщения о связи частых эпизодов ОГС с развитием хронического заболевания легких [33], не меньшее количество публикаций сообщает об отсутствии у пациентов с часто повторяющимися ОГС в будущем повреждения легких [31,33].
Инфаркт и некроз костного мозга – известные осложнения СКБ [36]. Когда инфаркт массивный, то некротизированный костный мозг и жир эмболизируют легочные сосуды [35,36]. Капельки жира могут попасть в системный кровоток, что приводит к развитию синдрома системной жировой эмболии (ССЖЭ) [37]. Таким образом, в дополнении к респираторной несостоятельности у пациентов развивается полиорганная недостаточность от эмболов в органах, например, почках и мозге. ССЖЭ может поражать пациентов даже с легким течением СКБ. Факторы риска развития ССЖЭ [37] – генотип HbSC, беременность, предшествующее лечение глюкокортикостероидами. Клинические проявления сильно варьируют и зависят от того, какой орган и в какой степени вовлечен. В начале – болевой криз с быстрым появлением лихорадки, гипоксемии, азотемии, поражение печени, изменение сознания или комы. Гематологические черты – прогрессирующая анемия, нормобластоз в периферической крови, тромбоцитопения, ДВС. ССЖЭ часто подозревается, но достаточно тяжело его подтвердить. Непрямые доказательства ССЖЭ – наличие макрофагов загруженных жировыми включениями в бронхоальвеолярном лаваже, жировые включения в клетках легочных микрососудов или наличие жировых включений в лейкомассе венозной крови или множественные очаги некроза костного мозга при сканировании. Поражение легких (ОГРС, жировая эмболия легких) часто предшествует или сопровождает ССЖЭ, таким образом начатая трансфузионная терапия гипоксемии у пациентов с ОГС останавливает или предотвращает развитие ССЖЭ [35–37].
Дисфункция печени и желчевыводящих путей – одно из наиболее частых осложнений СКБ [38]. Гепатобилиарные осложнения можно разделить на несколько категорий: связанные с гемолизом, вызванные анемией и ее трансфузионной терапией, последствия «серповидности» и вазо-окклюзии, заболевания несвязанные с СКБ [38].
Хронический гемолиз, с его ускоренным обменом билирубина, приводит к высокой заболеваемости желчнокаменной болезнью [38,39]. Как правило, только по причине гемолиза концентрация билирубина у пациентов СКБ не превышает 68 мкмоль/л, конъюгированная фракция составляет менее 10%. Заметное увеличение неконъюгированной фракции было зарегистрированы в связи с генетическим дефектом глюкуронилтрансферазной системы (синдром Жильбера) [40,41].
Ультразвуковое обследование популяций пациентов показало, что начало желчнокаменной болезни приходиться на возраст от 2 до 4 лет [42]. С возрастом частота встречаемости желчнокаменной болезни увеличивается, достигая к 18 годам 30%. Распространенность этого осложнения зависит от пищевых привязанностей пациентов (пациенты, имеющие в рационе питания преобладание растительных волокон, реже имеют холелитиаз) и генотипа заболевания. Ксенобиотики, такие как цефалоспорины 3-его поколения, могут кристаллизоваться в просвете желчного пузыря, а различия в использовании таких антибиотиков может объяснить некоторые географические различия в частоте развития холелитиаза. Сонаследование α-талассемии уменьшает степень гемолиза и, соответственно, частоту образования камней в желчном пузыре. Обструкция общего желчного протока часто неполная, поскольку пигментные камни небольшие, но они все же могут вызывать характерные биохимические изменения холестаза. Желчнокаменная болезнь может протекать с или без панкреатита [38,39,42–44].
Желчный осадок представляет собой вязкий материал, который не дает акустической тени на УЗИ и может быть предвестником развития желчного камня. Некоторые антибиотики, такие как цефтриаксон**, могут способствовать образованию осадка. Исследования у пациентов с СКБ показывают, что в желчном пузыре осадок часто встречается с камнями, но осадок сам по себе может и не прогрессировать в образование камней [42–44]. Тем не менее, период наблюдения за пациентами в таких исследованиях мал.
Лихорадка, тошнота, рвота и боли в животе – частые проявления при многих заболеваниях печени, кишечника, поджелудочной железы, позвонков, легких и неврологических заболеваний [45]. В таблице 1 представлен перечень заболеваний, которые могут в дебюте иметь вышеперечисленные симптомы, и часто встречаются у пациентов СКБ.
Таблица 1. Причины боли в животе (правый верхний квадрант) и изменения печеночных показателей в биохимическом анализе крови.

Необходимо проведение тщательного клинического обследования для установления четкого диагноза. Сцинтиграфия печени может быть полезной, но ее использование является спорным из-за высокого процента ложно положительных результатов и низкой прогностической ценности положительного результата. Тем не менее, она имеет высокую отрицательную прогностическую ценность. Ложно положительный результат может возникнуть в результате длительного голодания, тяжелых заболеваний печени, обструкции внепеченочной, хронического холецистита, или наркотически индуцированного спазма сфинктера Одди.
Описано несколько случаев прогрессирующего холестаза, которые характеризовались болью в верхнем правом квадранте живота, экстремально высоким содержанием билирубина, резким повышением щелочной фосфатазы, вариабельным повышением печеночных трансаминаз, часто сопровождались почечной недостаточностью, тромбоципенией и выраженной гипокоагуляцией. Смертность за счет неконтролируемой кровоточивости и печеночноклеточной недостаточности [46].
Секвестрация в селезенке – основная причина острого глубокого снижения гемоглобина, что не редко приводит к летальному исходу (у детей одна из основных причин смерти) [22,47].
Острая секвестрация в селезенке (ОСС) развивается в результате задержки эритроцитов в селезенке с последующим развитием стремительного снижения Hb (и высоким риском развития гипоксического шока. Диагностическими критериями ОСС можно считать стремительное снижение Hb на 20 г/л относительно исходного, резко выраженный ретикулоцитоз, остро развившаяся спленомегалия. ОСС может возникнуть в любом возрасте начиная с 5 недель жизни, наиболее часто с 3 мес. до 5 лет, часто провоцируется вирусной или бактериальной инфекцией. Обычные клинические проявления при ОСС – внезапная слабость, бледность, тахикардия, тахипноэ и увеличение живота. Смертность при ОСС составляет 20%. Прогностические факторы развития ОСС не определены. Наиболее часто и в более раннем возрасте ОСС развивается у пациентов с HbSS СКБ, реже при HbSC и S/β+-талассемией (средний возраст 9 лет) [47].
Почки у пациентов с СКБ имеют множественные структурные и функциональные нарушения на протяжении всего нефрона [48]. Мозговое вещество почки, место расположения почечных канальцев, сосудов (часто называется vasa recta) характеризуется гипоксией, ацидом, гипертоничностью, что способствует полимеризации HbS и, соответственно, серповидности эритроцитов, что приводит к быстрому нарушению кровоснабжения, а вслед за этим и функции. С возрастом отмечается резкое снижение количества сосудов в этой области, оставшиеся сосуды значительно расширены и спиралевидно извиты и часто слепо заканчиваются. В таблице 2 суммированы все почечные осложнения вызванные СКБ [48].
Таблица 2. Поражение почек при СКБ.


Гипостенурия – неспособность концентрировать мочу – наиболее частое почечное осложнение СКБ, которое развивается уже в раннем детском возрасте и проявляется в виде энуреза. Нарушение концентрационной функции почек часто сочетается с никтурией [48,49]. В результате нарушенной способности концентрировать мочу пациенты с СКБ более чувствительны к дегидратации, фактору, который провоцирует развитие вазо-окклюзии. Поэтому пациентам с СКБ очень важно употреблять много жидкости для компенсации ее потери за счет гипостенурии [49].
Ночной энурез может быть проявлением гипоксии головного мозга, что происходит за счет пролиферативной эндотелиопатии и может быть одним из проявлений «немых» инсультов у пациентов с HbSS. Если ночной энурез сохраняется у детей старше 1 года необходимо провести обследование ребенка для исключения патологии мочевыделительной системы, провести анализ питьевого режима ребенка (равномерно распределить жидкость в течение дня), беседу с родителями о необходимости контроля за мочеиспусканиями ребенка (будить ребенка ночью для дополнительного мочеиспускания). Если к 6 годам не удалось купировать ночной энурез, то необходимо проводить исследование sO2 в ночное время, рекомендовать дополнительную оксигенацию помещения [1–5,7,8], при отсутствии ответа на стандартные для купирования ночного энуреза рекомендации рассмотреть вопрос о терапии десмопрессином** [49].
У пациентов с СКБ часто встречается дисфункция канальцев [49]. Обычно пациенты имеют нормальный ответ на альдостерон и ренин. Первично – неполный ацидоз дистальных почечных канальцев, тяжесть нарушения кислотности мочи связана отчасти с тяжестью гипостенурии [49].
Нарушение экскреции калия не приводит к гиперкалиемии при сохранении функции почек, но при использовании пациентами антигипертензивных средств (ингибиторы АПФ, бета-адреноблокаторы и калийсберегающие диуретики) может развиться гиперкалиемия [1–5,7,8,49].
Повышение секреции креатинина приводит к низкому содержанию креатинина в сыворотке крови и, таким образом, переоценке функции клубочковой фильтрации у пациентов СКБ. Разница составляет до 30% при оценке скорости клубочковой фильтрации по клиренсу инулина. Эту особенность необходимо учитывать при назначении препаратов, у которых почки – основной путь выведения [49].
Несмотря на повышение секреции мочевой кислоты пациенты с СКБ часто имеют гиперурикемию и предрасположены к вторичной подагре [49].
Гематурия – частое осложнение при СКБ, которое развивается а результате полимеризации HbS и серповидности эритроцитов в мозговом слое почек [1–5,7,8]. Она может быть манифестным признаком папиллярного некроза или почечной медуллярной карциномы [49]. В большинстве случаев кровопотеря происходит из левой почки, крайне редко вовлекается билатеральная почка.
Гематурия может появиться и у носителей HbS (HbAS) [1–5,7,8,49].
ОПН развивается у пациентов с СКБ как одно из проявлений синдрома острой полиорганной недостаточности (СОПОН) [1–5,7,8,49]. Этот синдром характеризуется внезапным началом тяжелой дисфункции как минимум двух основных систем организма (например, почки, легкие, печень) в течение острого болевого вазо-окклюзивного криза. Патофизология СОПОН – вазо-окклюзия мелких сосудов в органах-мишенях с развитием ишемии паренхимы органов и нарушение их функции. ОПН может сопровождать рабдомиолиз.
Протеинурия, которая может прогрессировать в нефротический синдром, — наиболее частая манифестация поражения клубочков у пациентов СКБ [1–5,7,8,49]. Более того, до 40% пациентов СКБ (HbSS) могут перейти в хроническую почечную недостаточность (ХПН) [50]. Ингибиторы АПФ могут уменьшить патологические изменения такие, как фокальный или сегментарный гломерулосклероз, снизить экскрецию белка почками у пациентов с ранней манифестацией серповидноклеточной нефропатии. Почечная недостаточность более рано развивается у пациентов с HbSS, чем с HbSC. Предикторами развития ХПН у пациентов СКБ (HbSS) являются артериальная гипертензия, протеинурия, возрастающая тяжесть анемии и гематурия, кроме того, высокий риск развития ХПН отмечается у пациентов с «Центрально-Африканским» гаплотипом кластера βS-глобинового гена.
Приапизм – стойкая самопроизвольная болезненная эрекция [51]. Средний возраст развития этого осложнения 12 лет, к 20 годам 89% мужчин пациентов СКБ имели один или несколько эпизодов. Причина развития – вазо-окклюзия венозного дренажа полового члена. Приапизм считается длительным, если продолжается более трех часов, кратковременным, если его продолжительность от нескольких минут до трех часов и купируется самостоятельно [1–5,7,8,51]. Повторные эпизоды приапизма приводят к фиброзу и импотенции. Необходимо объяснить пациентам, что провоцировать развитие приапизма может полный мочевой пузырь, поэтому необходимо регулярное частое опорожнение мочевого пузыря; длительная сексуальная активность; наркотики и сильнодействующие препараты (например, кокаин, алкоголь, тестостерон, психолептики, силденафил); инфекция мочевыделительной системы, травма уретры [51].
Хронический гемолиз при СКБ приводит к гиперплазии эритроидного ростка костного мозга, что сопровождается деформацией костей, более заметной на голове. Нарушение роста верхней челюсти приводит к протрузии резцов и нарушению прикуса. В длинных костях остеопения предрасполагает к патологическим переломам костей. Аналогичные изменения наблюдаются и в телах позвонков, что сопровождается компрессионными переломами, приводящими к уплощению и кифотической деформации позвоночника. Протрузия головки бедренной кости с одной или двух сторон у пациентов с СКБ, также объясняется остеопенией вследствие эритроидной гиперплазии [52].
Аваскулярный некроз головок плечевой и бедренной костей может произойти при любом генотипе СКБ. Головка плечевой кости чаще повреждается у пациентов старшей возрастной группы. При сохраняющихся болях в плечевом и/или тазобедренном суставах необходимо проведение МРТ соответствующего сустава с определением радиологической стадии поражения [52,53].
Кости и суставы – основные места боли при вазо-окклюзионных кризах. Внезапный инфаркт сопровождается острой болью и другими симптомами, сходными с остеомиелитом. Наиболее часто инфаркты происходят в позвонках, костях таза, длинных костях (наиболее часто плечевая кость, берцовая кость, бедренная кость; указаны по мере снижения частоты поражения), при этом чаще поражаются дистальные отделы. Клинически отмечается – отечность, уплотнение и повышение температуры кожи над очагом, снижение объема движения в близлежащем суставе. В отличие от остеомиелита лихорадки как правило не наблюдается, редко может быть субфебрилитет, крайне редко отмечается левый сдвиг в лейкоцитарной формуле. Специфической рентгенологической картины нет, поэтому дифференцировать от острого остеомиелита весьма сложно [1–5,7,8,52].
Дактилит или синдром «руки-ноги» отмечается только у новорожденных и детей раннего возраста. Одновременно может быть поражена одна или все 4 конечности. Клинические проявления – боль в пястных, плюсневых и фаланговых костях рук и ног, отечность типично располагается по тыльной поверхности кисти и стопы, продолжаясь на пальцы. Рентгенологически отмечается утолщение надкостницы и «изъеденность молью» пораженной костью. Симптомы обычно проходят через 1-4 недели без каких-либо последствий [1–5,7,8,54].
Трофические язвы нижних конечностей у детей встречаются редко [1–6]. Трофические язвы как правило двухсторонние, локализованы в области лодыжек. Они могут быть как безболезненными, так и сопровождаться интенсивной болью. Патогенез до конца не ясен, наиболее вероятно они являются следствием нарушения микроциркуляции серповидными эритроцитами и низкой оксигенации тканей. Не исключается роль дефицита цинка в их развитии [1–8]. Терапия гидроксикарбамидом** или программной заместительной терапией эритроцитной массы не влияют на частоту развития и степень тяжести трофических язв.
Осложнения беременности у пациенток с СКБ (таблица 3) [55].
Таблица 3. Осложнения беременности у женщин, пациентов СКБ.

Диагностика
Диагностика заболевания или состояния (группы заболеваний или состояний), медицинские показания и противопоказания к применению методов диагностики
Диагноз СКБ устанавливается на основании клинических проявлений и данных лабораторного обследования [1–8], позволяющих выявить аномальный гемоглобин и/или поломки глобиновых генов. Методы лабораторной диганостики СКБ:
- общий (клинический) анализ крови с подсчетом ретикулоцитов и морфологической оценкой эритроцитов;
- гипоксическая проба на обнаружение серповидных эритроцитов,, электрофорез гемоглобина, изоэлектрофокусировка гемоглобина, капиллярный электрофорез гемоглобина, высокоэффективная жидкостная хроматография (методы расположены по возрастанию эффективности выявления аномального гемоглобина);
- ДНК-исследование глобиновых генов
Жалобы и анамнез
- Всем пациентам с подозрением на СКБ или с верифицированной СКБ при каждом посещении врача-гематолога рекомендуется сбор анамнеза и жалоб при заболеваниях органов кроветворения и крови [1–8].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: основная жалоба – частые эпизоды боли различной интенсивности и локализации, сопровождающиеся повышением температуры, бледность кожи и слизистых, в раннем возрасте могут быть головные боли с неврологическими проявлениями. Могут быть жалобы на энурез, нарушение зрения, одышку, желтуху.
Сбор анамнеза при СКБ подразумевает тщательный расспрос о возрасте появления первых симптомов заболевания, наличие в семье детей или взрослых с аналогичными проявлениями (заболеванием).
При осмотре (первичном или при динамическом наблюдении) необходимо документировать анамнестические данные:
— клинические проявления СКБ на момент осмотра, какие и какой степени тяжести эпизоды (болевые кризы, недомогания и т.п.) были за прошедшее с момента последнего осмотра время;
— наличие/отсутствие головных болей, приапизма, болей в животе, ночного энуреза, каких-либо неврологических проявлений, которые могли бы свидетельствовать о возможном инсульте;
— выполняются ли рекомендации по пенициллинопрофилактике;
— соблюдаются ли рекомендации по вакцинопрофилактике, как переносились вакцинации;
— как купируют боль и лихорадку дома;
— как часто пропускает ребенок школу и по какой причине;
— тесты для оценки когнетивных функций, успеваемость в школе;
— какие физические упражнения выполняются ребенком и/или посещение каких спортивных секций;
— планируются ли поездки (если да, то куда).
Физикальное обследование
- Всем пациентам с подозрением на СКБ или с верифицированной СКБ при каждом посещении врача-гематолога рекомендуются визуальное исследование, пальпация и перкуссия при заболеваниях органов кроветворения и крови [1–6].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: общий осмотр подразумевает оценку общего физического состояния, роста и массы тела, наличия вторичных половых признаков в соответствующем возрасте, рост, масса тела, физическое развитие; степень выраженности бледности, желтухи, размеры печени и селезенки, наличие шума в сердце; артериальное давление.
СКБ у детей раннего возраста имеет различные проявления. Самым первым клиническим проявлением может быть желтуха в течение первых нескольких недель жизни. Если гемолиз не интенсивный, то о наличии желтухи свидетельствует желтушный цвет склер. У детей первых месяцев жизни часто встречается гепатомегалия. Часто при СКБ (все формы, кроме СКА) выявляется увеличенная селезенка. При длительно существующей анемии у пациентов СКБ может выслушиваться мезосистолический шум при аускультации сердца. В более старшем возрасте обращает на себя внимание отставание в физическом и половом развитии [1–8].
Лабораторные диагностические исследования
- Всем пациентам с подозрением на СКБ или с верифицированной СКБ при каждом посещении врача-гематолога рекомендуется выполнение общего (клинического) анализа крови развернутого, дифференцированного подсчета лейкоцитов (лейкоцитарная формула), исследование уровня ретикулоцитов в крови, просмотр мазка крови для оценки состояния пациента и для анализа аномалий морфологии эритроцитов, тромбоцитов и лейкоцитов [1–6,56,57].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
- Всем пациентам с подозрением на СКБ или с верифицированной СКБ каждые 3-6 месяцев рекомендуется выполнение биохимического анализа крови общетерапевтического (общий билирубин, креатинин, мочевина, АЛТ, АСТ) для оценки функции печени и почек [58].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
- Всем пациентам с подозрением на СКБ рекомендуется выполнение следующих исследований для выявления аномального гемоглобина (в порядке возрастания эффективности выявления) [1–6,56,57]:
— гипоксическая проба на обнаружение серповидноклеточных эритроцитов (только у детей старше 2 лет, т.к. присутствие большого количества HbF делает его ложно отрицательным);
— определение соотношения белковых фракций (гемоглобина) методом электрофореза;
— выявление типов гемоглобина;
— определение соотношения белковых фракций (гемоглобина) методом высокочувствительного капиллярного электрофореза;
— комплексное определение концентрации на аминокислоты методом высокой эффективной жидкостной хроматографии
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
- Всем пациентам с подозрением на СКБ рекомендуется выявление точечных мутаций в гене глобина (ПЦР с последующим рестрикционным анализом, секвенирование, аллель-специфическая амплификация для выявления делеционной альфа-талассемии) для выявления генетических аномалий, приведших к развитию СКБ [1–6,56,57].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Инструментальные диагностические исследования
- Всем пациентам с СКБ начиная с 2-летнего возраста ежегодно рекомендуется дуплексное сканирование брахиоцефальных артерий с цветным допплеровским картированием кровотока для раннего выявления инсульта [1–6,59–62].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3)
Комментарии: у пациентов детского возраста с увеличением скорости кровотока по средней мозговой артерии более 200 см/сек риск развития инсульта в ближайшие 3 года возрастает на 40%, поэтому начиная с 2-х летнего возраста ежегодно необходимо проведение транскраниального дуплексного (допплеровского) исследования (ТКИ) кровотока по сосудам головы; при скорости кровотока по средней мозговой артерии менее 170 см/сек исследование повторяется через 1 год, при скорости кровотока 170-199 см/сек необходимо повторить исследование через 1-4 месяца, при сохранении результата и при возрасте пациента старше 10 лет необходимо проведение МРТ
- Всем пациентам с СКБ начиная с 2-летнего возраста при невозможности проведения дуплексного сканирования брахиоцефальных артерий и при выявлении отклонений от нормальных значений скорости кровотока по магистральным сосудам головы рекомендуется магнитно-резонанская томография (МРТ) головного мозга с контрастным усилением или в режиме ангиографии [1–6,59–62].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3)
- Всем пациентам с подозрением на СКБ рекомендуется МРТ костной ткани или (при невозможности проведения МРТ) ультразвуковое исследование костей для выявления остеонекроза и аномалий костей и суставов [1–6,53,63].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
- Пациентам с СКБ с малыми субарахноидальными кровоизлияниями рекомендуется выполнение церебральной ангиографии для выявления аневризмы или артериовенозной мальформации для возможного хирургического вмешательства [1,2,64–68].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
- Всем пациентам с СКБ, получающим регулярные трануфузии эритроцитарной массы, рекомендуется МРТ сердца с контрастированием, МРТ органов брюшной полости с внутривенным введением гепатотропного контрастного препарата для диагностики перегрузки железом [69].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: МРТ в режиме Т2 – наиболее информативный метод в диагностике перегрузки железом
Иные диагностические исследования
- Всем пациентам с СКБ, начиная с раннего возраста, рекомендуются ежегодный осмотр врачом-офтальмологом и расширенное офтальмологическое обследование для диагностики офтальмологических осложнений СКБ [1,2,7,8,70].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: пациенты с СКБ при травме глаза или периорбитальной области должны быть немедленно осмотрены офтальмологом, т. к. они имеют высокий риск потери зрения от повышения внутриглазного давления с кровоизлиянием в переднюю камеру (гифема).
Лечение
Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
Цель лечения – обеспечить нормальный рост и развитие ребенка, сохранить трудоспособность в подростковом возрасте, обеспечить хорошее качество жизни пациента
1. Консервативное лечение
- Всем пациентам с СКБ старше 2 лет при наличии тяжелых болевых кризов, требующих госпитализации 3 и более раз в год либо 2 или более эпизодов острого грудного синдрома рекомендуется физиологическая стимуляция синтеза HbF #гидроксикарбамидом** внутрь в режиме 15-35 мг/кг/сут (округляя в большую сторону до целой капсулы) ежедневно постоянно [1,2,67,71–73,3–7,64–66].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: постоянный длительный прием #гидроксикарбамида** обеспечивает у 90% пациентов существенное повышение содержания HgF, достаточное для существенного улучшения течения СКБ.
Критерии гематологического ответа:
1. полный — Hb >100 г/л, отсутствие болевых кризов и других осложнений СКБ;
2. частичный — Hb 85-100 г/л, отсутствие значимых болевых кризов и других осложнений СКБ;
3. отсутствие ответа — Hb <85 г/л, частые болевые кризы различной интенсивности, могут быть и другие осложнения СКБ.
#Гидроксикарбамид** экскретируется почками, поэтому у пациентов c нарушением функции почек необходимо более внимательно мониторировать дозу #гидроксикарбамида**.
Пациенты, получающие #гидроксикарбамид**, должны использовать адекватную контрацепцию. В случае принятия решения о рождении ребенка #гидроксикарбамид** должен быть отменен за 1 и более месяц до зачатия ребенка [1,2,74–76]. Есть описания случаев, когда #гидроксикарбамид** не был отменен и принимался пациентками в течение всей беременности, пороков развития плода отмечено не было. Методами выбора могут быть пероральные гормональные контрацептивы (G03A по АТХ классификации) или парентеральные (внутримышечные) гормональные контрацептивы для системного применения – гестагены (L02AB по АТХ классификации), барьерная контрацепция [1,2,77]. Использование внутриматочных контрацептивов не рекомендуется в связи с высоким риском тяжелых осложнений, связанных с СКБ [1,2,77].
2. Трансфузионная терапия эритроцитарной массой
Трансфузии эритроцитной массы – эффективный метод лечения острых, потенциально летальных случаев анемии и тяжелых вазо-оклюзивных кризов. Режим трансфузий зависит от лечебного плана и осложнений, приведших к началу трансфузионной терапии.
В 18% случаев трансфузии у пациентов СКБ осложняются аллоиммунизацией, для снижения риска развития необходимо трансфузировать эритроцитную массу совместимую по группе крови, резус-фактору и Kell-антигену.
- Пациентам с СКБ рекомендуется простая малообъемная трансфузия эритроцитарной массы (взвеси) (<7 мл эр.массы/кг, целевой Hb 100-110 г/л) в следующих клинических ситуациях [78–83]:
— острый грудной синдром;
— нарушение функции любого органа;
— сиквестрация в селезенке или печени;
— сепсис или малярия;
— рефрактерный болевой вазо-окклюзивный криз;
— острая анемия с клиническими проявлениями анемического синдрома;
— предоперационная подготовка.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Пациентам с СКБ рекомендуется объемная трансфузия эритроцитарной массы (взвеси) (концентрация HbS в крови пациента не должна превышать 30%, целевой Hb 120-130 г/л) в следующих клинических ситуациях [78–83]:
— инсульт;
— недавняя полная потеря слуха;
— тромбоз центральной артерии сетчатки;
— подготовка к обширным хирургическим вмешательствам (грудная клетка, сердце, глаза и т.п.);
— острый рефрактерный приопизм.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Пациентам с СКБ рекомендуются хронические регулярные трансфузии эритроцитарной массы (взвеси) (концентрация HbS в крови пациента не должна превышать 30%, целевой Hb 120-130 г/л) в следующих клинических ситуациях [78–83]:
— церебральная васкулопатия;
— повторный тяжелый острый грудной синдром и/или тяжелый болевой (вазо-оклюзивный) криз;
— хроническая органная недостаточность: почки, сердце, легкие, печень;
— легочная гипертензия;
— тяжелая задержка роста;
— 3-й триместр беременности;
— психоз;
— тяжелая хроническая анемия, не отвечающая на терапию гидроксикарбамидом**.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Пациентам с СКБ при развитии синдрома гипергемолиза рекомендуется пульс-терапия метилпреднизолоном** в сочетании с #иммуноглобулином человека нормальным** (в/в введение – код АТХ J06BA02) в курсовой дозе 0,4 г/кг/сут [84–86].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: частота развития отсроченных гемолитических посттрансфузионных реакций (синдром гипергемолиза) у пациентов с СКБ составляет 4-22%, что существенно выше, чем при других заболеваниях. При развитии синдрома гипергемолиза в некоторых случаях аллоантител обнаружить не удается.
- Пациентам с СКБ после 10-15 трансфузий эритроцитной массы (взвеси) при ферритине сыворотки не менее 1000 мкг/л для поддержания ферритина сыворотки в диапазоне 800-1000 мкг/л рекомендуется хелаторная терапия деферазироксом** (лекарственая форма таблетка диспергируемая — начальная доза 30 мг/кг/сут внутрь ежедневно, далее с шагом 5 мг/кг/сут повышается или понижается в зависимости от ферритина сыворотки; лекарственная форма таблетка, покрытая пленочной оболочкой, — начальная доза 21 мг/кг/сут внутрь ежедневно, далее с шагом 3,5 мг/кг/сут повышается или понижается в зависимости от ферритина сыворотки) [87].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: отмена хелаторной терапии при достижении сыворотки 600 мкг/л. При проведении хелаторной терапии необходимо контролировать:
1. уровень железа сыворотки крови, ОЖСС/НЖСС, насыщения трансферрина железом ( НТЖ), уровень ферритина в крови каждые 3 мес. при подборе дозы хелатора, далее каждые 6 мес.;
2. клиренс эндогенного креатинина до начала хелаторной терапии, каждые 3 мес. на этапе подбора дозы, далее каждые 6-12 мес.;
3. МРТ Т2* печени и миокарда 1 раз в год.
3. Трансплантация гемопоэтических стволовых клеток
- Пациентам с СКБ моложе 17 лет в случае наличия родственного HLA-совместимого донора и при развитии одного из осложнений СКБ (инсульт (или МРТ признаки инсульта без клинических проявлений), церебральная васкулопатия со снижением когнитивной функции, тяжелый или повторный острый грудной синдром, хроническая дыхательная недостаточность 1-2 ст., 3 и более болевых (вазо-оклюзивных) криза, требовавших госпитализации, в год в течение последних 3-4 лет) рекомендуется трансплантация гемопоэтических стволовых клеток (ТГСК) [1,2,88–90].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: показано, что после родственной HLA-совместимой ТГСК выживаемость без возврата СКБ составляет 80-92%, смертность, в основном от хронической РТПХ, составляет 5-10% [90–92].
- Пациентам с СКБ не рекомендуется ТГСК в следующих случаях [90]:
— индекс Карновского <70%;
— портальный фиброз средней или выраженной степени;
— нарушение клубочковой функции почек средней или тяжелой степени;
— выраженное нарушение интелекта;
— множественный эпифизарный аваскулярный некроз;
— хроническая дыхательная недостаточность 3-4 ст.;
— кардиомиопатия;
— ВИЧ инфекция;
— донор является пациентом с гемоглобинопатией.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
4. Хирургическое лечение
Пациенты с СКБ могут нуждаться в хирургических вмешательствах как для лечения осложнений СКБ, так и в связи с несвязанными с СКБ состояниями. Риск развития осложнений и риск смерти от анестезии у таких пациентов существенно выше, чем в общей популяции, в основном за счет присутствия анемии, способности эритроцитов превращаться в серповидные и закупоривать просвет мелких сосудов и капилляров, наличие хронического повреждения органов у некоторых пациентов, усугубления имеющейся гипоксии и наконец эффекта асплении. Наиболее высоки риски у пациентов с HbSS и S/β0-талассемией [1,2,66,67,93,3–8,64,65]. Проведено много клинических исследований, направленных на снижение риска хирургических вмешательств и анестезии. Частота развития послеоперационных осложнений увеличивается с возрастом пациента (~1,3 на каждые 10 лет жизни) [1,2,64–67,93,94]. Осложнения развиваются чаще у пациентов, которым проводили региональную анестезию, нежели при общей анестезии [1,2,93,94]. Аденотомия и тонзиллэктомия у пациентов с СКБ расцениваются как оперативные вмешательства высокого риска в связи с кровопотерей, невозможностью пить, потерей жидкости [1,2,64–67,93,94].
Во время предоперационной подготовки необходимо обратить внимание на гематокрит, периферическую перфузию и кислородный статус [1,2,64–67,93–96]. Минимум за 8 часов до оперативного вмешательства необходимо предоперационое введение жидкостей (гемоделюция) и вне зависимости от степени риска оперативного вмешательства проведение трансфузии эритроцитной массы: при низком риске – простая трансфузия для обеспечения Hb ~100 г/л, при среднем и высоком – обменная (снижение концентрации HbS менее 30%) [1,2,64–67,93]
Интраоперационно нужно мониторировать АД, ритм и частоту сердечных сокращений, оксигенацию, и температуру (предупреждение охлаждения) на протяжении всего оперативного вмешательства [1,2,64–67,93,94].
Послеоперационное ведение – обеспечение дыхания кислородом с контролем газов крови (sO2), гидратация и репираторная поддержка/терапия [1,2,64–67,93,94].
Частые хирургические вмешательства при СКБ – аденотонзиллэктомия, спленэктомия, холецистэктомия (обычно лапароскопическая), замена купных суставов, установка порта инфузионного/инъекционного, имплантируемого [1,2].
- Пациентам с СКБ с подтвержденной аневризмой или артериовенозной мальформацией рекомендуется выполнение хирургического лечения (иссечение аневризмы или удаление артериовенозной мальформации) [1,2,64–68].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
- Пациентам с СКБ с отслойкой сетчатки или обширным кровоизлиянием в стекловидное тело рекомендуется хирургическое вмешательство – витреоэктомия [1,2,70].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: хотя современная витреоретинальная микрохирургия может улучшить зрение для многих пациентов с поздними стадиями ретинопатии при СКБ, следует подчеркнуть, что операция несет в себе существенный риск интраоперационных и послеоперационных осложнений, в том числе тяжелой ишемии глаза, повторных кровоизлияний и повышения внутриглазного давления. Для уменьшения подобного риска перед оперативным вмешательством необходимо проведение частичного заменого переливания крови (HbA должен составить в кровотоке пациента минимум 50-60%)
- Пациентам с СКБ с окклюзией центральной артерии сетчатки рекомендуется экстренное (не позднее 1 часа от момента возникновения) вмешательство – оксигенотерапия в комбинации с быстрым снижением внутриглазного давления (хирургически и терапевтически) [1,2,70].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Пациентам с СКБ с потерей зрения вследствие кровоизлияния или отслойки сетчатки рекомендуется экстренное (не позднее 48 часов от момента возникновения) хирургическое вмешательство [1,2,70].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Пациентам с СКБ с показаниями к холецистэктомии рекомендуется выполнение лапароскопической холецистэктомии с эндоскопической ретроградной холангиопанкреатографией [1,2,95,96].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: лапароскопическая холецистэктомия для хорошо подготовленного пациента – стандартный подход. Агрессивный подход обеспечивает преимущество снижения риска осложнений желчно-каменной болезни, а также устранение болезни желчного пузыря в дифференциальной диагностике боли в правом верхнем квадранте живота.
4. Профилактика осложнений СКБ
- У пациентов с СКБ при развитии лихорадки неясного генеза рекомендуется быстрое реагирование и агрессивное лечение, включающее [66,97–99]:
— госпитализацию в случае подъема температуры выше 38,5С;
— базовое обследование, включающее: общий анализ крови, общий анализ мочи, рентгенографию органов грудной клетки, исследование кислотно-основного состояния и газов крови, микробиологическое (культуральное) исследование крови на стерильность, микробиологическое (культуральное) исследование слизи с миндалин и задней стенки глотки на аэробные и факультативно-анаэробные микроорганизмы, микробиологическое (культуральное) исследование мочи на бактериальные патогены с применением автоматизированного посева;
— незамедлительное (до готовности результатов обследования) назначение антибактериальных препаратов парентерально в случае наличия у ребенка симптомов интоксикации и/или лихорадки выше 40,0С, либо детям без симптомов интоксикации с температурой ниже 40,0С в следующих клинических ситуациях:
изменение sO2 или инфильтративные изменения на рентгенограмме органов грудной клетки;
число лейкоцитов более 30х109/л или менее 5х109/л;
число тромбоцитов менее 100х109/л;
гемоглобин менее 50 г/л;
сепсис в анамнезе;
— проведение люмбальной пункции с микрбиологическим (культутальным) исследованием спинномозговой жидкости на аэробные и факультативно-анаэробные условно-патогенные микроорганизмы в случае наличия симптомов интоксикации с менингиальными симптомами
— дети могут получать лечение в дневном стационаре, если у них нет интоксикации при температуре менее 40С, никогда не было сепсиса, рентгенография органов грудной клетки или кислотно-основное состояние и газы крови без патологии, пограничное количество лейкоцитов, тромбоцитов и гемоглобин; они могут быть отпущены под амбулаторное наблюдение после назначения парентеральной антибактериальной терапии активной против Streptococcus pneumonia и Hemophilus influenzae (например, цефтриаксон** 75 мг/кг) при условии, что дети остаются клинически стабильными в течение 3 часов после введения антибиотика, эндемичный в регионе Streptococcus pneumonia чувствителен к назначенному антибиотику, родители обучены наблюдению за пациентом СКБ, четко выполняют рекомендации врача, назначенное в дневном стационаре лечение будет продолжено по месту жительства в амбулаторном режиме.
— документированная бактериемия должна быть пролечена парентерально в течение 7 дней, дети с менингитом должны получить не менее 14 дней терапии.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: после 3 мес. жизни при существенном снижении HbF появляется риск развития гипофункции селезенки с повышением восприимчивости к некоторым жизни угрожающим бактериальным инфекциям (сепсису, менингиту и др.). Исследования, проведенные в США, показывают, что при HbSS резко возрастает частота тяжелых бактериальных инфекций, включая менингит и сепсис в течение первого полугодия жизни, при HbSC и S-β+-талассемии в течение первых 2-х лет жизни [1–5,100–106]. Важное место занимает профилактика инфекций (пенициллинопрофилактика и вакцинация), поэтому необходимо контролировать четкость и своевременность ее проведения вне зависимости от возраста пациента.
Фебрильные пациенты с СКБ должны рассматриваться в свете функциональной асплении при обследовании и лечении. Это подразумевает, что в кратчайшие сроки таким пациентам необходимо провести физикальный осмотр, сделать общий (клинический) анализ крови, микробиологические (культуральные) исследования крови на стерильность и/или слизи с миндалин и задней стенки глотки на аэробные и факультативно-анаэробные микроорганизм и/или мочи на бактериальные патогены с применением автоматизированного посева, рентгенографию легких, и начать эмпирическую антибактериальную терапию. У детей с СКБ часто бывает незначительный фибрилитет, а риск летального исхода от недиагностированного Streptococcus pneumonia сепсиса колоссален.
- Детям с СКБ раннего возраста, начиная с 2-месячного возраста, рекомендуется пеницилллинопрофилактика [1,2,107–109,3–5,64–68].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Всем пациентам с СКБ рекомендуется регулярная санация очагов инфекции (зубная полость, миндалины, аденоиды, придаточные пазухи носа, мочевыводящая система) [1,2].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: очаги хронической инфекции как правило провоцируют развитие тяжелых вазо-окклюзивных кризов, могут быть причиной сепсиса.
- Всем пациентам с СКБ вне зависимости от возраста рекомендуется гиперкалорийная диета, что позволяет существенно улучшить темпы роста [1,2,7,8,64–68,110–112].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: у пациентов с СКБ, особенно детей, отмечается задержка роста, низкий нутритивный статус, задержка скелетного и полового созревания. Задержка роста становиться заметной начиная с 6 месячного возраста. Считается, что это связано с ускорением метаболизма при СКБ, снижением аппетита (иногда до анорексии) при болевых кризах, снижении абсорбции питательных веществ в ЖКТ. Многочисленные исследования пациентов СКБ показали снижение содержания жировой ткани в подростковом возрасте, снижение содержания нежировых компонентов в структуре тела у всех пациентов вне зависимости от возраста, при этом существенно снижена мышечная масса и белковые запасы организма [1,2,7,8,64–68,110–112].
- Пациентам с СКБ с повышением концентрации гомоцистеина в крови рекомендуется назначение препаратов группы «фолиевая кислота и ее производные» (В03ВВ по АТХ классификации) для коррекции их концентрации [1,2,113–115].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Всем пациентам с СКБ рекомендуется профилактика дефицита витамина Д: дотация колекальциферола**, достаточное пребывание пациента на солнце и адекватное содержание кальция в рационе питания [116,117].
Уровень убедительности рекомендаций A (уровень достоверности доказательств – 1)
- Пациентам с СКБ при отсутствии признаков пубертата у девочек в возрасте 14 лет и у мальчиков в возрасте 14,5 лет рекомендуется прием (осмотр, консультация) врача-эндокринолога первичный для решения вопроса о необходимости гормонозаместительной терапии [1,2,7,8,64–68].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: у пациентов с HbSS отмечается задержка пубертата на 2-3 года, у пациентов с HbSC – на ~6 месяцев. Задержка созревания скелета в период пубертата продляет период роста длинных костей, что позволяет достичь приемлемый конечный рост пусть и с задержкой во времени.
- Всем пациентам с СКБ с проявлениями приапизма рекомендуются инъекции #лейпрорелина** (аналог гонадотропин-рилизинг гормона, который угнетает гипоталамо-тестикулярную связь и продукцию тестостерона) 7,5 мг в/м, длительная терапия #гидроксикарбамидом** внутрь в режиме 15-35 мг/кг/сут (округляя в большую сторону до целой капсулы) ежедневно постоянно для предотвращения повторных эпизодов приапизма [1,2,7,8,64–68,118].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: подростки и взрослые пациенты СКБ должны быть оповещены о том, что при сохранении признаков приапизма в течение 2-х и более часов должны обратиться за неотложной помощью к врачу. Для купирования легких проявлений и профилактики развития тяжелого приапизма пациенты должны всегда перед сном опорожнять мочевой пузырь, для купирования легких эпизодов – принять препарат из группы «анальгетики и антипиретики» (N02BG по АТХ классификации) и горячую ванну.
5. Профилактика поражения ЦНС
Помимо изменения скорости кровотока по средней мозговой артерии, к факторам риска относятся [1,2,7,8,119–122]:
— транзиторные ишемические атаки (транзиторное нарушение мозгового кровообращения);
— низкий гемоглобин;
— частота и время с момента развития острого грудного синдрома;
— повышенное систолическое давление.
Для детей первых двух лет жизни факторами высокого риска развития ишемического инсульта являются дактилит, тяжелая анемия, лейкоцитоз [1,2,7,8,119–122].
К другим клиническим и лабораторным индикаторам риска развития ишемического инсульта относят: инсульт у родного брата/сестры, мелкие неврологические отклонения, тяжелую анемию, высокий лейкоцитоз, определенные βS-гаплотипы и отсутствие делеции α-глобиновых генов [1,2,7,8,119].
Примерно 13% детей (до 20% пациентов до 20-летнего возраста) с СКБ имеют «немое» поражение мозга, по данным МРТ очаги локализуются преимущественно в лобной и теменной долях в корковых, подкорковых и пограничной зонах. Эти повреждения ассоциированы с низкой оценкой при нейропсихологическом тестированием. Доказано повышение риска развития ишемического инсульта у детей, которые имеют отклонения по данным МРТ [1,2,7,8,119]. Присутствие этих поражений при МРТ требует проведение обследования ребенка для выявления проблем обучения, когнитивных нарушений и оценки сосудов головного мозга для своевременного начала первичной профилактики инсульта. Выявленное «немое» поражение головного мозга у ребенка требует переоценки истории болезни для выявления ранее не замеченных симптомов, а также пересмотр клинических и лабораторных рисков развития инсульта. Частота инсульта у детей с измененным МРТ и ТКИ, а также риски и преимущества профилактически хроническими трансфузиями эритроцитной массы не исследованы. В настоящее время проводятся исследования по изучению эффективности регулярных трансфузий эритроцитной массы при «немых» инсультах.
- Пациентам с СКБ с повышением скорости кровотока по средней мозговой артерии до 200 см/сек рекомендуется проведение регулярных (хронических) трансфузий эритроцитарной массы для предупреждения развития ишемического инсульта [1,2,7,8,120,121].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: хронические трансфузии эритроцитной массы со снижением содержания HbS менее 30% от общего Hb значительно снижают риск повторного инсульта [1,2,7,8,120]. Хронические трансфузии эритроцитной массы должны продолжаться до 18-летнего возраста ребенка, но не менее 5 лет от начала трансфузий [1,2,7,8,120].
Некоторые пациенты, несмотря на адекватное снижение HbS трансфузиями эритроцитной массы, развивают повторные эпизоды поражения ЦНС. В этом случае необходимо определить содержание HbS в крови пациента (убедиться, что трансфузионная программа у пациента была адекватной), выяснить причину развития ишемии. Необходимо помнить о других факторах риска, кроме обусловленной СКБ васкулопатии: повышение содержания гомоцистеина и другие состояния, сопровождающиеся гиперкоагуляцией. Повышение гомоцитеина может быть редуцировано приемом фолиевой кислоты**. Есть данные, что некоторые пациенты с СКБ имеют повышенное содержание антифосфолипидных антител и дефицит протеина С и S [1,2,7,8,120,123–125].
- Детям с СКБ с гиперкоагуляцией рекомендуется проведение антикоагулянтной терапии варфарином** [126,127].
Уровень убедительности рекомендаций B (уровень достоверности доказательств – 3)
- Пациентам с СКБ, перенесшим инфаркт/ишемию могза, рекомендуется проведение трансплантации гемопоэтических стволовых клеток для предотвращения повторных атак [1,2,7,8,88,89,92].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Взрослым пациентам с СКБ рекомендуется проведение профилактики и лечения ишемического инсульта по педиатрическим клиническим рекомендациям [1,2,7,8].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Взрослым пациентам с СКБ с транзиторной ишемической атакой (ТИА) рекомендуется проведение профилактики ишемического инсульта антитромботическими средствами или варфарином** [1,2,7,8,128–130].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: препараты для вторичной профилактики инсульта у взрослых пациентов с СКБ представлены в таблице 5 [131,132].
Таблица 5. Препараты для вторичной профилактики инсульта у взрослых пациентов СКБ.


В качестве альтернативы у взрослых пациентов с СКБ для вторичной профилактики ишемического инсульта могут применяться хронические (регулярные) трансфузии эритроцитарной массы аналогично педиатрической практике [1,2,7,8,82]. Хирургические вмешательства могут быть использованы в некоторых случаях для коррекции синдрома Моямоя [1,2,7,8].
- Взрослым пациентам с СКБ рекомендуется вторичная проведение профилактики ишемического инсульта ацетилсалициловой кислотой** или варфарином** [1,2,7,8,128–130].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
1. Болевые кризы при СКБ
Боль при СКБ можно подразделить на острую и хроническую [1–7,64–68,134–154]. Состояния, ассоциированные с болевыми кризами представлены в таблице 7. Наиболее часто при СКБ встречается острая боль, ее начало без каких-либо предвестников, продолжительность ее ограничена: от нескольких часов до нескольких дней, интенсивность боли существенно варьирует от умеренной до невыносимой, боль может возвращаться, локализация боли может варьировать. Хроническая боль – боль, продолжающаяся 3-6 месяцев и более, трудно дифференцируемая от часто повторяющейся острой боли (например, боль при поражении костей), может быть изнурительной, как физически, так и психологически. Участие ощущений, эмоций, памяти и восприятия создают дополнительные проблемы в ее купировании. Часто острая боль может накладываться на хроническую, а частые приступы острой боли могут напоминать хроническую боль. Нейропатическая боль недостаточно охарактеризована при СКБ, она возникает в результате инфарктов нервных структур, сдавления, действия эндотоксинов и/или перегрузки железом.
Таблица 7. Острая и хроническая боль и осложнения СКБ [1,2,67,68,138–140,142,144,146,148,3–7,64–66].

В оценке интенсивности боли необходимо учитывать возраст пациента, уровень его развития, функциональный статус, когнитивную функцию, эмоциональное состояние. Все эти факторы должны также учитываться при выборе терапии. Купирование боли должно быть быстрым и максимально эффективным [1,2,67,68,134–141,3,142,4–7,64–66].
В купировании боли большую роль играют образование родителей и пациента в области патологии СКБ и причин развития боли [1,2,140–142,64–68,137–139]. Очень важно для купирования тяжелых эпизодов боли наличие дневных стационаров в зоне досягаемости для пациентов, что снижает количество и продолжительность госпитализаций в круглосуточный стационар [1,2,134,136,139,140].
Необходимо избегать факторов, провоцирующих болевые кризы, таких как: воздействие низкой температуры (охлаждение), воздействие высокой температурой (перегрев), длительная интенсивная физическая нагрузка, дегидратация, подъем на высоту более 1500 м над уровнем моря, табакокурение, употребление алкогольных напитков [1,2,140–142,64–68,137–139].
Эффективно использование немедикаментозных способов купирования боли (согревающий массаж, теплый/горячий душ или ванна, рефлексотерапия, отвлекающие мероприятия (игры, в т. ч. компьютерные, видео; TV) [1,2,140–142,64–68,137–139].
На рис. 1 представлен алгоритм оценки интенсивности и характера боли у пациентов с СКБ [1,2].
Рисунок 1. Алгоритм оценки интенсивности и характера боли у пациентов с СКБ


- Для оценки интенсивности боли у детей рекомендовано использовать визуальную шкалу оценки боли (см. приложение Г1 данных рекомендаций) или другие мультимерные шкалы для острой или хронической боли [1,2,140–142,64–68,137–139]. Ведение дневника пациента также помогает в оценке боли дома.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Купирование боли у пациентов с СКБ должно проводиться согласно соответствующим клиническим рекомендациям.
2. Транзиторная красноклеточная аплазия
Транзиторная красноклеточная аплазия (ТКА) развивается вследствие инфицирования парвовирусом В19 (парвовирус В19 также вызывает развитие erythema infectiosum, известную как «пятая болезнь»). Аплазия является результатом прямого цитотоксического действия парвовируса на эритроидные предшественники, в какой-то степени могут повреждаться и предшественники других клеточных линий. Пациенты могут иметь возрастающую головную боль, слабость, диспноэ, более тяжелую чем обычно анемию и глубокое снижение числа ретикулоцитов (обычно менее 1% или 10 х 109/л). Также может быть лихорадка, признаки инфекции верхних дыхательных путей и/или гастроинтестинальные симптомы. Кожные высыпания не имеют специфических характеристик. Ретикулоцитопения появляется примерно на 5 день заражения и продолжается в течение 5-10 дней. Утяжеление анемии происходит вскоре после ретикулоцитопении, Hb снижается до 39 г/л. Первый признак начала выздоровления от инфекции – высокий ретикулоцитоз, что при сохранении глубокой анемии иногда ошибочно трактуется как синдром гипергемолиза. Выздоровление как правило сопровождается появлением в периферической крови большого числа нормобластов (более 100 на 100 лейкоцитов). Диагноз ТКА подтверждается повышенным содержанием IgM к парвовирусу В19 в крови. При выздоровлении от парвовирусной В19 инфекции появляется защитный титр IgG, что препятствует повторному заболеванию этой инфекцией в течение всей жизни пациентов.
Контролируемых исследований по терапии ТКА не проводилось. Часть пациентов выздоравливают самостоятельно.
Хотя большинство взрослых приобрели иммунитет к парвовирусу B19, работники медицинской организации, которые восприимчивы и имеют контакт с пациентами ТКА подвергаются высокому риску внутрибольничного заражения инфекционной эритемой (erythema infectiosum). Перенесенная во втором триместре беременности инфекция может привести к водянке плода и мертворождению, поэтому необходимы изоляционные меры предосторожности для персонала в случае беременности.
- Пациентам с глубокой ТКА рекомендованы трансфузии эритроцитарной массы [1,2,64–68].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: наиболее часто трансфузии требуются у пациентов с HbSS, наименее часто – HbSC.
- Пациентам с ТКА рекомендована терапия #иммуноглобулином человека нормальным** в лекарственной форме «раствор для инфузий» (ВВИГ**) в дозе 2 г/кг в/в, разделенной на 5 дней [155].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
3. Поражение глаз при СКБ
Периферическая вазо-окклюзия сосудов сетчатки может быть выявлена уже в возрасте 20 месяцев жизни, клинически значимое поражение сетчатки обычно отмечается в возрасте 15-30 лет. Серповидно-клеточная» ретинопатия наиболее часто и рано развивается у пациентов HbSC, но и у пациентов HbSS и S-β-талассемией бывают также достаточно часто. Стадия IV и V ретинопатии чаще диагностируется у пациентов HbSC, чем у пациентов HbSS. Менее тяжелое системное поражение при СКБ у пациентов с HbSC и S-β-талассемией сопровождается парадоксально более тяжелым поражением глаз, чем при HbSS. Риск развития пролиферативной ретинопатии увеличивается с возрастом [1–6].
При выявлении пролиферативной ретинопатии пациент должен быть направлен в специализированный офтальмологический центр.
- В целях диагностики пролиферативной ретинопатии у пациентов с СКБ рекомендованы осмотр с расширенным зрачком с использованием широко-полосного непрямого офтальмоскопа (щелевая лампа), оценка кровотока в сетчатке методом флюоресцентной ангиографии [1,2,70].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Пациентам с СКБ и пролиферативной ретинопатией в случаях двухсторонней пролиферативной ретинопатии, спонтанных кровоизлияниях, больших расширенных ветвей новых сосудов, стремительной неоваскуляризации, и когда один глаз уже серьезно пострадал от пролиферативной ретинопатии, рекомендованы раннее лечение с использованием следующих методов — диатермии, криотерапии, лазерной коагуляции в целях регрессии неоваскуляризации ткани до развития кровотечения и отслойки сетчатки. Терапевтическое вмешательство обычно необходимо. [1,2,70].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
4. Сердечно-сосудистые осложнения СКБ
Хроническая сердечная недостаточность у пациентов СКБ должна лечиться по соответствующим клиническим рекомендациям. Глубокая анемия в сочетании с хронической сердечной недостаточностью или стенокардией нуждается в компенсации (регулярные трансфузии эритроцитной массы или терапия гидроксикарбамидом**) [156].
- Начать антигипертензивную терапию у пациентов с СКБ рекомендовано в следующих случаях [1,2,28,157]:
— при повышении систолического АД на 20 мм рт. ст. или диастолического АД на 10 мм рт. ст.
— при АД выше 130/85 мм рт. ст., если выявлено поражение сердца, нефропатия или васкулопатия периферических сосудов;
— при АД 120/75 мм рт. ст. в случае протеинурии (более 1 г/сут).
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: Ингибиторы ангиотензин превращающего фермента или антагонисты кальция особенно эффективны [1,2,28,64–68]. Диуретики следует использовать крайне осторожно, т.к. они могут спровоцировать развитие вазоокклюивныого криза [1,2,28,64–68]. Возможно использование бета-адреноблокаторов [1,2,28]. Тяжелое повышение артериального давления требует исключения вторичной гипертензии.
5. Острый грудной синдром
Терапия гидроксикарбамидом** снижает частоту развития ОГС на 50% [1,2,28,31,32]. Регулярная трансфузионная терапия также может снизить частоту развития ОГС [1,2,28,31,32].
- При развитии острого грудного синдрома (ОГС) у пациентов с СКБ рекомендовано выполнить исследование кислотно-основного состояния и газов крови и рассчитывать альвеолярно-артериальный градиент кислорода и отношение РаО2/FiO2 для оценки тяжести состояния пациента [1,2,31–35,64–68,158–160].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: При ухудшении альвеолярно-артериального градиента кислорода необходим перевод пациента в отделение интенсивной терапии и реанимации для обеспечения адекватной кардиореспираторной поддержки.
- Пациентам с СКБ и ОГС с умеренной гипоксемией (РаО2=70-80 мм рт. ст., sО2=92-95%) рекомендовано назначение кислорода интраназально со скоростью 2 л/мин [32–35].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Контроль боли в груди и стимуляционная спирометрия рекомендованы всем пациентам с СКБ и ОГС, т.к. могут предотвратить развитие гиповентиляции у большинства пациентов [1,2,31–35,64–68,158–160].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- При развитии ОГС рекомендовано провести трансфузию эритроцитной массы (простую или обменную) [1,2,31–35,64–68,158–160]. Основное показание для трансфузии в этом случае – низкая дыхательная функция; цель – предотвратить прогрессию ОГС в острую дыхательную недостаточность. Трансфузионная терапия начинается при первых признаках гипоксемии ((РаО2 ниже 70 мм рт ст при дыхании комнатным воздухом), для пациентов с хронической гипоксемией снижение РаО2 на 10% и более от базового значения.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Трансфузионная терапия не показана в случае, если отмечена положительная динамика альвеолярно-артериального градиента кислорода при адекватном обезболивании и стимуляционной спирометрии. Трансфузионную терапию не следует задерживать, особенно при ухудшении пациента. Измененный процесс мышления у таких пациентов часто ошибочно приписывается избытку опиоидов, что приводит к отсрочке терапии прогрессирующего ОГС.
- Внутривенное назначение антибактериальных препаратов широкого спектра действия настоятельно рекомендовано при наличии лихорадки или тяжелой степени тяжести ОГС у пациентов с СКБ, т.к. достаточно сложно исключить бактериальную пневмонию или суперинфекцию инфаркта легкого [1,2,31–35,64–68,158–160]. Рекомендуется использовать эритромицин и препараты из группы других бета-лактамных антибиотиков (J01D по АТХ классификации). В комбинации антибактериальной терапии всегда должен присутствовать препарат группы макролидов или фторхинолонов, т.к. атипичные микроорганизмы весьма часто присутствуют.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Рекомендовано при развитии гиперреактивности бронхов (возникает у одной четверти пациентов с ОГС) назначение селективных бета 2-адреномиметиков [1,2,28,31,32].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
6. Острая секвестрация в печени
Острая секвестрация в печени, редко диагностируемое осложнение вазо-окклюзивного криза, характеризуется стремительно нарастающим увеличением размеров печени в сочетании с быстрым снижением гематокрита/Hb и повышением числа ретикулоцитов. При пальпации печень гладкая, плотность варьирует. Общий билирубин может быть существенно повышенным с преобладанием коньюгированной фракции, повышение активности щелочной фосфатазы до 650 МЕ/л (иногда бывает в пределах нормы), печеночные трансаминазы как правило в пределах нормы или слегка повышены. УЗИ и КТ показывают диффузное увеличение печени, при гистологическом исследовании – резко расширенные синусы, заполненные серповидными эритроцитами и эритрофагоцитоз купферовскими клетками. Может быть внутрипеченочный холестаз, некроз гепатоцитоз не типичен. Выздоровление происходит весьма часто.
- В тяжелых случаях острой секвестрации в печени у пациентов с СКБ рекомендовано обменное переливание эритроцитной массы (при простой трансфузии очень высокий риск резкого повышения вязкости крови и общего объема циркулирующей крови при разрешении секвестрационного криза), в средне тяжелых и легких случаях – симптоматическая терапия [1,2,38,45,161].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
7. Острая секвестрация в селезенке
- При развитии острой секвестрации в селезенке (ОСС) у пациентов с СКБ рекомендовано незамедлительное начало лечения, оно должно быть начато немедленно (ОСС может быть фатальным в течение первых часов), главное — коррекция гиповолемии путем трансфузии эритроцитной массы [1,2,22,47,64–68,133,162–165].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: После трансфузии эритроцитарной массы эритроциты «задержанные» селезенкой освобождаются, вместе с этим уменьшаются размеры селезенки и повышается Hb (причем в большинстве случаев выше, чем ожидается с учетом трансфузии). ОСС может повторяться, что зависит от дальнейшего лечения пациента: динамическое наблюдение, хронические (регулярные) трансфузии эритроцитной массы, спленэктомия (тотальная) [1,2,22,47,64–68,133,162–166].
- Хронические (регулярные) трансфузии эритроцитной массы рекомендуются детям с СКБ до 2-летнего возраста [1,2,22,47,64–68,162,163].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- Всем пациентам СКБ в возрасте старше 2 лет при развитии тяжелого ОСС рекомендуется проведение спленэктомии [1,2,22,47,64–68,133,162–166].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
8. Гематурия при СКБ
- При развитии у пациентов с СКБ гематурии, после исключения злокачественного заболевания почек [167], рекомендованы следующие мероприятия:
— постельный режим,
— контроль диуреза (выпито/выделено),
— при существенной кровопотере – назначение препаратов железа или трансфузии эритроцитной массы [1,2,168,169],
— возможна терапия вазопрессином и его аналогами (H01BA02 по АТХ классификации) и аминокапроновой кислотой** (6-8 г/сут в 4-6 приемов, для детей – в соответствии с возрастом согласно инструкции, позволяет купировать гематурию в течение 2-3 дней) [1,2,169]. Данная комбинация может приводить к тромбообразованию в сосудах почек, которое разрешается самостоятельно в течение 2-37 дней, поэтому ее назначение должно производиться с осторожностью.
— при длительном и жизни-угрожающем кровотечении из почки -хирургическое лечение: резекция кровоточащего сегмента или односторонняя нефрэктомия [1,2,168].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
9. Ночной энурез при СКБ
- Если к 6 годам у пациентов с СКБ не удалось купировать ночной энурез, то провести определение степени насыщения кислородом гемоглобина в ночное время, рекомендовать дополнительную оксигенацию помещения [1,2,170,171].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
10. Ведение беременности и родов при СКБ
- Пациентам (пациенткам) с СКБ, планирующим рождение ребенка, а также родителям ребенка с СКБ рекомендовано генетическое консультирование для определения риска рождения больного СКБ ребенка либо вероятности рождения еще одного больного СКБ ребенка в данной семье при последующих беременностях.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- При возникновении беременности пациенткам с СКБ необходимо раннее обращение к акушеру-гинекологу для проведения стандартного обследования с целью минимизировать обычные риски развития осложнений беременности. Вместе с тем, учитывая наличие СКБ, рекомендовано дополнительно провести следующее обследование [1,2,172]: электрофорез/ВЭЖХ Hb; обмен железа (сывороточное железо, ОЖСС, ферритин сыворотки) [172].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- В случае неосложненного течения беременности у пациентки с СКБ рекомендуется воздержаться от трансфузионной терапии. В случае развития осложнений (преэклампсия, анемия с уровнем гемоглобина ниже 90 г/л, учащение болевых эпизодов), а также начиная с 20 недели у пациенток с многоплодной беременностью или с выкидышем или мертворожденными в предыдущей беременности рекомендовано начать регулярные трансфузии эритроцитарной массы.
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Требование к эритроцитарной массе – тщательная лейкодиплеция, индивидуальный подбор с учетом минорных групп крови. Если во время беременности необходимо снизить содержание HbS, а Hb высокий, то необходимо сначала удалить 500 мл цельной крови, затем трансфузировать 2 дозы эритроцитарной массы. Это можно проводить как вручную, так и на сепараторе клеток, при этом посттрансфузионный Hb должен быть 100-110 г/л, а HbS 30-40% от общей концентрации Hb [1,2,172–175].
- Не рекомендовано прерывание беременности у пациентов с СКБ на сроке более 13 недель, т.к. сопряжено с большими рисками для женщины [1,2,176].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Прерывание беременности у пациенток с СКБ возможно на сроке до 13 недель, обеспечив достаточное обезболивание [1,2,176]. На сроке более 13 недель прерывание беременности сопряжено с большими рисками для женщины.
Роды у пациенток с СКБ.
Сердечная функция может быть скомпрометирована хронической гипоксией и анемией у большинства пациенток.
- При необходимости операции Кесарева сечение перед операцией рекомендовано провести трансфузию эритроцитной массы (простую или заменую) [1,2,172].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
- В послеродовом периоде рекомендована заместительная трансфузия эритроцитной массы пациенткам с СКБ при при большой кровопотери [1,2,172]. Часто в послеродовый период у женщин с СКБ осложняется венозной тромбоэмболией, для ее профилактики также рекомендовано в ранние сроки расширять двигательную активность пациенток [1,2,172].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Медицинская реабилитация
Медицинская реабилитация и санаторно-курортное лечение, медицинские показания и противопоказания к применению методов медицинской реабилитации, в том числе основанных на использовании природных лечебных факторов
Специфических реабилитационных мероприятий в отношении пациентов с СКБ не разработано. Пациенты с СКБ вне зависимости от возраста и получаемой терапии могут посещать детские дошкольные, школьные учреждения, пребывать в оздоровительных лагерях, заниматься физической культурой и спортом. Пациенты, перенесшие инсульт, особенно повторный, будут нуждаться в социальной реабилитации.
Пациентам с СКБ полезно заниматься физкультурой и спортом, однако при этом необходимо помнить о необходимости сохранения тепла (после интенсивных физических занятий ребенка необходимо укрыть/одеть в теплое для предотвращения переохлаждения; не рекомендуется плавать в прохладной/холодной воде) и контролировать прием ребенком достаточного количества жидкости для предотвращения дегидратации, что будет способствовать предотвращению развития вазо-окклюзивного криза.
Госпитализация
Организация оказания медицинской помощи
Показания для госпитализации в медицинскую организацию:
1. плановая госпитализаци в дневной стационар (стационар кратковременного лечения):
a. проведение трансфузий эритроцитной массы (взвеси)
2. плановая госпитализация в стационар:
a. предоперационная подготовка
b. 3-й триместр беременности
3. экстренная госпитализация в дневной стационар (стационар кратковременного лечения):
a. болевой синдром средней и тяжелой степени (рефрактерный болевой вазо-окклюзивный криз)
4. экстренная госпитализация в стационар:
a. острый грудной синдром
b. секвестрация в селезенке или печени
c. сепсис или малярия
d. острая анемия с клиническими проявлениями анемического синдрома
e. инсульт
f. острый рефрактерный приопизм
Показания к выписке пациента из медицинской организации
1. достижение целевого гемоглобина
2. купирование болевого синдрома и разрешение осложнения или сопутствующего заболевания.
Профилактика
Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
После установления диагноза, выбора лечебной тактики, подбора доз препаратов пациент передается под диспансерное наблюдение врача-педиатра (если есть должность, то под наблюдение гематолога) по месту жительства. Терапия проводится амбулаторно, длительно/пожизненно. Пациенты и члены их семей должны быть подробно ознакомлены как с сутью заболевания, возможным осложнениям как самого заболевания, так и проводимой терапии, обучены правилам индивидуальной гигиены и купированию боли.
- Всем пациентам с СКБ рекомендуется пожизненное наблюдение врача-гематолога и врача-педиатра/терапевта с привлечением при необходимости других специалистов [1,2,64–68].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: в большинстве своем пациенты нуждаются в амбулаторном лечении и/или профилактике тяжелых проявлений заболевания или его осложнений, крайне редко требуется лечение в стационаре.
Необходимые мероприятия при диспансерном наблюдении представлены в таблице 6.
Таблица 6. Необходимые медицинские услуги при диспансерном наблюдении [1,2,7,8,65–68,133]






Передача во взрослую сеть медицинских учреждений
должна быть постепенной. Планирование и подготовка передачи пациента под наблюдение должно быть начато по достижении ребенком возраста 13-14 лет [1,2]; детальная выписка из всей медицинской документации пациента должна быть подготовлена к 15-16 годам [1,2]; гематолог из взрослого/подросткового центра должен принимать участие в обсуждении пациента, начиная с 13-14 лет, что позволяет обеспечить преемственность в оказании медицинской помощи и знакомство ребенка с правилами работы центра, который будет в дальнейшем его наблюдать [1,2].
Информация
Источники и литература
-
Клинические рекомендации Национального гематологического общества
- 1. Sickle cell disease in childhood. Standards and guidelines for clinical care. 2nd edition. 2010. 88 p.
2. The Management of Sickle Cell Disease. National Heart, Lung, and Blood Institute (NHLBI), 2002. 206 p.
3. Rees D.C., Williams T.N., Gladwin M.T. Sickle-cell disease // The Lancet. 2010. Vol. 376, № 9757. P. 2018–2031.
4. Stuart M.J., Nagel R.L. Sickle-cell disease // Lancet. 2004. Vol. 364, № 9442. P. 1343–1360.
5. Steinberg M.H. In the clinic. Sickle cell disease // Annals of internal medicine. 2011. Vol. 155, № 5.
6. Румянцев А.Г., Токарев Ю.Н., Сметанина Н.С. Гемоглобинопатии и талассемические синдромы. М.: Практическая медицина, 2015. 448 p.
7. Costa F.F., Conran N. Sickle cell anemia: From basic science to clinical practice // Sickle Cell Anemia: From Basic Science to Clinical Practice. Springer International Publishing, 2016. 1–435 p.
8. Serjeant G.R. One hundred years of sickle cell disease // British Journal of Haematology. 2010. Vol. 151, № 5. P. 425–429.
9. Malowany J.I., Butany J. Pathology of sickle cell disease // Semin. Diagn. Pathol. 2012. Vol. 29, № 1. P. 49–55.
10. Vos T. et al. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013 // Lancet. Lancet Publishing Group, 2015. Vol. 386, № 9995. P. 743–800.
11. Naghavi M. et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013 // Lancet. Lancet Publishing Group, 2015. Vol. 385, № 9963. P. 117–171.
12. Fernandes A.P.P.C. et al. Mortality of children with sickle cell disease: a population study // J Pediatr (Rio J). 2010. Vol. 86, № 4. P. 279–284.
13. Scheinin L., Wetli C. V. Sudden death and sickle cell trait: Medicolegal considerations and implications // Am. J. Forensic Med. Pathol. 2009. Vol. 30, № 2. P. 204–208.
14. Platt O.S. et al. Mortality in sickle cell disease — life expectancy and risk factors for early death // N. Engl. J. Med. 1994. Vol. 330, № 23. P. 1639–1644.
15. Wierenga K.J., Hambleton I.R., Lewis N.A. Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study. // Lancet (London, England). 2001. Vol. 357, № 9257. P. 680–683.
16. Quinn C.T., Rogers Z.R., Buchanan G.R. Survival of children with sickle cell disease // Blood. 2004. Vol. 103, № 11. P. 4023–4027.
17. Serjeant G.R., Higgs D.R., Hambleton I.R. Elderly survivors with homozygous sickle cell disease // New England Journal of Medicine. Massachussetts Medical Society, 2007. Vol. 356, № 6. P. 642–643.
18. Yanni E. et al. Trends in Pediatric Sickle Cell Disease-Related Mortality in the United States, 1983-2002 // J. Pediatr. 2009. Vol. 154, № 4. P. 541–545.
19. Quinn C.T. et al. Improved survival of children and adolescents with sickle cell disease // Blood. 2010. Vol. 115, № 17. P. 3447–3452.
20. Makani J. et al. Mortality in Sickle Cell Anemia in Africa: A Prospective Cohort Study in Tanzania // PLoS One / ed. Schrijver I. 2011. Vol. 6, № 2. P. e14699.
21. Darbari D.S. et al. Circumstances of death in adult sickle cell disease patients // Am. J. Hematol. 2006. Vol. 81, № 11. P. 858–863.
22. Khatib R., Rabah R., Sarnaik S.A. The spleen in the sickling disorders: An update // Pediatric Radiology. 2009. Vol. 39, № 1. P. 17–22.
23. Ohene-Frempong K. et al. Cerebrovascular accidents in sickle cell disease: Rates and risk factors // Blood. 1998. Vol. 91, № 1. P. 288–294.
24. Leight L. et al. Hemodynamic studies in sickle cell anemia. // Circulation. 1954. Vol. 10, № 5. P. 653–662.
25. Covitz W. et al. The heart in sickle cell anemia: The cooperative study of sickle cell disease (CSSCD) // Chest. American College of Chest Physicians, 1995. Vol. 108, № 5. P. 1214–1219.
26. Braden D.S., Covitz W., Milner P.F. Cardiovascular function during rest and exercise in patients with sickle-cell anemia and coexisting alpha thalassemia-2 // Am. J. Hematol. 1996. Vol. 52, № 2. P. 96–102.
27. Johnson C.S., Giorgio A.J. Arterial blood pressure in adults with sickle cell disease. // Arch. Intern. Med. 1981. Vol. 141, № 7. P. 891–893.
28. Pegelow C.H. et al. Natural history of blood pressure in sickle cell disease: Risks for stroke and death associated with relative hypertension in sickle cell anemia // Am. J. Med. 1997. Vol. 102, № 2. P. 171–177.
29. Martin C.R. et al. Myocardial infarction in sickle cell disease. // J. Natl. Med. Assoc. 1996. Vol. 88, № 7. P. 428–432.
30. Romero Mestre J.C. et al. Cardiovascular autonomic dysfunction in sickle cell anemia: a possible risk factor for sudden death? // Clin. Auton. Res. 1997. Vol. 7, № 3. P. 121–125.
31. Paul R.N. et al. Acute chest syndrome: Sickle cell disease // European Journal of Haematology. 2011. Vol. 87, № 3. P. 191–207.
32. Howard J. et al. Guideline on the management of acute chest syndrome in sickle cell disease // Br. J. Haematol. 2015. Vol. 169, № 4. P. 492–505.
33. Vichinsky E.P. et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group.[Erratum appears in N Engl J Med 2000 Sep 14;343(11):824] // N. Engl. J. Med. 2000. Vol. 342, № 25. P. 1855–1865.
34. Vichinsky E.P. et al. Acute chest syndrome in sickle cell disease: Clinical presentation and course // Blood. 1997. Vol. 89, № 5. P. 1787–1792.
35. Vichinsky E. et al. Pulmonary fat embolism: a distinct cause of severe acute chest syndrome in sickle cell anemia. // Blood. 1994. Vol. 83, № 11. P. 3107–3112.
36. Milner P.F., Brown M. Bone marrow infarction in sickle cell anemia: correlation with hematologic profiles. // Blood. 1982. Vol. 60, № 6. P. 1411–1419.
37. Castro O. Systemic fat embolism and pulmonary hypertension in sickle cell disease. // Hematol. Oncol. Clin. North Am. 1996. Vol. 10, № 6. P. 1289–1303.
38. Johnson C.S. et al. Liver involvement in sickle cell disease // Med. (United States). 1985. Vol. 64, № 5. P. 349–356.
39. Walker T.M., Hambleton I.R., Serjeant G.R. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. // J. Pediatr. 2000. Vol. 136, № 1. P. 80–85.
40. Stewart West M. et al. Laboratory profile of sickle cell disease: A cross-sectional analysis // J. Clin. Epidemiol. 1992. Vol. 45, № 8. P. 893–909.
41. Passon R.G., Howard T.A., Zimmerman S.A. The effect of UDP-glucuronosyltransferase (UGTIA) promotor polymorphisms on serum bilirubin levels and cholelithiasis in patients with sickle cell anemia // Blood. 1999. Vol. 94, № Supp. 1. P. 645a.
42. Nzeh D.A., Adedoyin M.A. Sonographic pattern of gallbladder disease in children with sickle cell anaemia. // Pediatr. Radiol. 1989. Vol. 19, № 5. P. 290–292.
43. Lee S.P., Maher K., Nicholls J.F. Origin and fate of biliary sludge // Gastroenterology. 1988. Vol. 94, № 1. P. 170–176.
44. Al-Salem A.H., Qaisruddin S. The significance of biliary sludge in children with sickle cell disease // Pediatr. Surg. Int. 1998. Vol. 13, № 1. P. 14–16.
45. Serafini A.N. et al. Diagnostic Studies in Patients With Sickle Cell Anemia and Acute Abdominal Pain // Arch. Intern. Med. 1987. Vol. 147, № 6. P. 1061–1062.
46. Shao S.H., Orringer E.P. Sickle cell intrahepatic cholestasis: approach to a difficult problem. // Am. J. Gastroenterol. 1995. Vol. 90, № 11. P. 2048–2050.
- 1. Sickle cell disease in childhood. Standards and guidelines for clinical care. 2nd edition. 2010. 88 p.
Информация
Список сокращений
АВМ – артериовенозная мальформация
АЛТ – аланинаминотрансфераза
АСТ – аспартатаминотрансфераза
АЧТВ – активированное частичное тромбопластиновое время
ВВИГ – препараты из группы иммуноглобулина нормального человеческого в лекарственной форме для внутривенного введения (Immunoglobulin normal human, for intravascular administration, J06BA02 по АТХ классификации)
ВИЧ – вирус иммунодефицита человека
ДВС – диссеминированное внутрисосудистое свертывание
ДНК – дезоксирибонуклеиновая кислота
ЖКТ – желудочно-кишечный тракт
КТ – компьютерная томография
ЛГ – легочная гипертензия
ЛДГ — лактатдегидрогеназа
МНО – международное нормализованное отношение
МР-ангиография – магнитрорезонанская ангиография
МРТ – магнитнорезонансная томография
НЖСС – ненасыщенная железосвязывающая способность сыворотки
НПВП – нестероидные противовоспалительные препараты
НТЖ – насыщение трансферина железом
ОГС – острый грудной синдром
ОЖСС – общая железосвязывающая способность
ОПН – острая почечная недостаточность
ОСС – острая секвестрация в селезенке
ПВ – протромбиновое время
ПЦР – полимеразная цепная реакция
РТПХ – реакция трансплантат против хозяина
САК – субарахноидальное кровоизлияние
СКБ – серповидно-клеточная болезнь
СОПОН – синдром острой полиорганной недостаточности
ССЖЭ – синдром системной жировой эмболии
ТГСК – трансплантация гемопоэтических клеток
ТИА – транзиторные ишемические атаки
УЗИ – ультразвуковое исследование
ХПН – хроническая почечная недостаточность
ЦНС – центральная нервная система
ЭКГ – электрокардиография
ЭХО-КГ – эхо-кардиография
AB0 – группа крови по системе AB0
ASA – ацетилсалициловая кислота**
Ig – иммуноглобулин
ER-DR – дипиридамол с замедленным высвобождением в комбинации с ацетилсалициловой кислотой (лекарственный препарат Агренокс, в РФ зарегистрирован без указания МНН)
Hb – гемоглобин
HbF – фетальный гемоглобин
HbSC – двойное гетерозиготное (компаунд-гетозиготное) носительство двух аномальных гемоглобинов HbS и HbC
HbSS – серповидно-клеточная анемия (гомозиготность по аномальному HbS)
HLA – главный комплекс гистосовместимости
MCV – средний корпускулярный объем эритроцита
PaO2 – парциальное давление кислорода
PLT – тромбоциты
Rh – резус-фактор
sO2 – сатурация кислорода
S-β-талассемия – двойная гетерозигота по аномальному HbS и β-талассемии (дрепаноталассемия)
t-PA – тканевой активатор плазминогена
TV – телевидение
WBC – лейкоциты
** – жизненно необходимые и важнейшие лекарственные препараты
# – препарат, применяющийся не в соответствии с показаниями к применению и противопоказаниями, способами применения и дозами, содержащимися в инструкции по применению лекарственного препарата (офф-лейбл)
Термины и определения
Аваскулярный некроз – ишемический некроз фрагмента кости, то есть некроз, возникающий в результате недостаточности кровоснабжения
Аденотомия – хирургическая операция удаления аденоидов
Аллоиммунизация – выработка антител к антигенам других людей
Анемия – снижение содержания гемоглобина
Аспления – отсутствие селезенки
Вакцинопрофилактика – специфическая профилактика инфекционных болезней путем вакцинации
Гепатомегалия – увеличение размеров печени
Гиперплазия эритроидного ростка костного мозга — увеличение числа структурных элементов тканей путём их избыточного новообразования
Гипостенурия – неспособность концентрировать мочу
Дактилит – заболевание, характеризующееся течением воспалительного процесса в пальце или пальцах кисти или стопы
Дезокси-форма гемоглобина – форма гемоглобина, в которой он способен присоединять кислород или другие соединения
Инсульт – острое нарушение мозгового кровообращения, сопровождающееся внезапной потерей сознания и параличами
Кифотическая деформация – искривление позвоночника в сагиттальной плоскости с образование выпуклости обращенной кзади
Контрацепция – предупреждение беременности с помощью противозачаточных средств
Лапароскопия — современный метод хирургии, в котором операции на внутренних органах проводят через небольшие (обычно 0,5—1,5 см) отверстия
Нейтропения – снижение числа нейтрофилов в периферической крови.
Никтурия – преобладание ночного диуреза над дневным
Нормохромная анемия – анемия с нормальным цветовым показателем (нормальным MCH) эритроцитов.
Остеомиелит – инфекционное воспаление всех составляющих частей костной ткани: кости, надкостницы и костного мозга
Остеонекроз – некроз костной ткани в результате плохого притока крови
Остеопения – состояние костной ткани, характеризующееся снижением минеральной плотности костной ткани, что приводит к хрупкости костей и повышенному риску их перелома
Острый грудной синдром – острые боли в грудной клетке, связанные с инфарктом легкого вследствие окклюзии сосудов при серповидно-клеточной анемии
Парциальная красноклеточная аплазия — заболевание или синдром, который клинически и лабораторно представлен глубокой анемией и избирательной (чистой) аплазией только красного ростка кроветворения
Пенициллинопрофилактика – применение антибиотиков пенициллинового ряда для профилактики инфекций, возбудители которых чувствительны к ним Пренатальная диагностика – комплексная дородовая диагностика с целью обнаружения патологии на стадии внутриутробного развития
Приапизм – непроизвольная стойкая эрекция, не связанная с сексуальным возбуждением
Пролиферативная ангиопатия – нарушение проницаемости кровеносных сосудов, обусловленное утолщением эндотелия сосудов
Протрузия – первая стадия формирования грыжи межпозвоночного диска
Рабдомиолиз – синдром, характеризующийся разрушением клеток мышечной ткани, резким повышением концентрации креатинкиназы и миоглобина, миоглобинурией, развитием острой почечной недостаточности
Серповидно-клеточная болезнь – группа состояний, обусловленных наличием специфических мутаций в бета-глобиновом гене, приводящих к изменению физико-химических свойств гемоглобина (полимеризация в деокси-форме, нарушение сродства к кислороду)
Спленэктомия – хирургическая операция по удалению селезёнки
Тонзиллэктомия – хирургическая операция, заключающаяся в удалении миндалин
Транзиторные ишемические атаки – транзиторное нарушение мозгового кровообращения
Трофические язвы – длительно не заживающий дефект кожи, возникающий в результате нарушения кровоснабжения этого участка кожи
Хелатор – вещество, образующее устойчивое нетоксичное соединение с металлом (в данном случае с железом), способное покинуть организм
Хелаторная терапия – использование хелаторов с лечебной целью
Холелитиаз – образование камней (конкрементов) в жёлчном пузыре, жёлчных протоках
Холестаз – патологический (ненормальный) процесс, связанный с нарушением синтеза (образования), секреции (выделения) и поступления желчи или ее отдельных компонентов в 12-перстную кишку
Холецистэктомия – операция по удалению желчного пузыря
Энурез – недержание мочи
Эритробластопения – снижение числа эритробластов в костном мозге
ASPEN синдром – сочетание СКБ, приапизма, заменной трансфузии и неврологического осложнения
Критерии оценки качества медицинской помощи


Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
Сметанина Наталия Сергеевна — доктор медицинских наук, профессор, член Национального общества детских гематологов и онкологов, член Национального гематологического общества, член Европейского общества гематологов, член Международного общества Биожелезо
Спиридонова Елена Александровна – доктор медицинских наук, член Национального общества по изучению шока
Цветаева Нина Валентиновна — кандидат медицинских наук, член Национального гематологического общества
Стефанов Дмитрий Николаевич – член Российского общества онкогематологов
Конфликт интересов
Сметанина Наталия Сергеевна – лектор, ООО «Новартис Фарма»
Спиридонова Елена Александровна – конфликт интересов отсутствует
Цветаева Нина Валентиновна – конфликт интересов отсутствует
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
1. Врач-гематолог
2. Врач-педиатр
3. Врач-терапевт
4. Врач общей практики
5. Врач-акушер-гинеколог
6. Врач-хирург
7. Врач-оториноларинголог
8. Врач-офтальмолог
9. Врач-анестезиолог-реаниматолог
10. Врач-трансфузиолог
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)

Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)


Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)

Порядок обновления клинических рекомендаций.
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию – не реже чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утверждённым КР, но не чаще 1 раза в 6 месяцев.
Приложение А3. Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата
Нет
Приложение Б. Алгоритмы действий врача


Приложение В. Информация для пациента
Серповидно-клеточная анемия – это генетически обусловленное заболевание, при котором происходит изменение формы эритроцитов при передаче кислорода в ткани, в результате чего меняется форма эритроцитов, эритроциты в этот момент могут закупоривать мелкие, иногда и средние, артерии, что сопровождается резкой болью в области остановки подачи крови, повышением температуры, вместе с тем эритроциты разрушаются, что приводит к снижению содержания гемоглобина (анемии) и пожелтению. Первые проявления такого заболевания могут быть как в первые месяцы жизни, так и в любом другом возрасте. Чем раньше проявилась болезнь, тем тяжелее она протекает.
Единственным способом излечить такое заболевание является трансплантация гемопоэтических клеток, что может быть проведено не всем пациентам. В большинстве случаев пациенты с серповидно-клеточной анемией получают лекарство для повышения фетального (плодового) гемоглобина, чтобы предотвратить тяжелые проявления заболевания, в тяжелых случаях, при хирургических вмешательствах или при развитии осложнений проводятся переливания донорской эритроцитной массы для снижения доли аномального гемоглобина в кровотоке. Вследствие болезни может исчезать селезенка, что сопровождается воспреимчивостью к некоторым инфекциям, что может привести к смерти, поэтому очень важно своевременно проводить профилактическую вакцинацию, и по назначению врача принимать специальные антибиотики. Важно регулярно посещать врача-педиатра (врача-терапевта) и/или врача-гематолога, которые будут контролировать лечение вашего заболевания, предупреждая развитие осложнений.
Важно помнить, что пациентов серповидно-клеточной болезнью нуждаются в гиперкалорийном питании, достаточном количестве жидкости (особенно в жаркое время года и при физической активности) и в физических упражнениях (занятии спортом); нельзя допускать переохлаждения, т.к. это может спровоцировать болевой криз.
При появлении первого болевого криза обязательно посоветуйтесь со своим врачом как быстро и эффективно снимать боль, в каких случаях обращаться за неотложной помощью.
При повышении температуры обязательно обратитесь к врачу сразу!
При появлении одышки на фоне повышения температуры или при невозможности остановить боль в течение 2-х часов немедленно обратитесь за неотложной помощью.
При выполнении всех необходимых мероприятий по лечению и контрольному обследованию продолжительность жизни и качество жизни пациентов соответствует средним значениям по популяции, существенных ограничений в выборе профессии нет.
Поскольку заболевание генетически обусловлено необходимо проведение ДНК-исследования генов, повреждение которых приводит к развитию данного заболевания. В дальнейшем это позволит провести пренатальную диагностику и предотвратить рождение
Приложение Г1 — ГN. Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведенные в клинических рекомендациях
Приложение Г1. Шкала для самооценки интенсивности боли для детей
Название на русском языке: «Шкала самооценки интенсивности боли для детей»
Оригинальное название (если есть):
Источник (официальный сайт разработчиков, публикация с валидацией): Wong D. et al. Whaley and Wong’s Nursing Care of Infants and Children, ed. 6. St. Louis, Mosby, 1999. 1153 p. [177]
Тип: шкала оценки
Содержание, ключ (интерпретация):

Прикреплённые файлы
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники. Стандарты лечения
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Внимание!
Если вы не являетесь медицинским специалистом:
-
Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
-
Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
-
Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
-
Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
-
Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Что такое серповидноклеточная анемия?
Серповидноклеточная анемия (СКА) — редкое заболевание крови, передающееся по аутосомно-рецессивному типу. СКА характеризуется наличием в кровотоке серповидных эритроцитов (красные кровяные тельца). Эти клетки в форме полумесяца жесткие и липкие и взаимодействуют с другими клетками и системой свертывания крови, блокируя кровоток в очень крошечных кровеносных сосудах (капиллярах) периферической кровеносной системы (кровеносных сосудах за пределами сердца). Это препятствует нормальному потоку питательных веществ и кислорода (поскольку красные кровяные тельца несут ответственность за перенос кислорода по всему телу).
Общие симптомы, связанные с СКА, включают мучительную боль в костях, боль в груди, тяжелые инфекции (в основном у детей), низкий уровень циркулирующих эритроцитов (анемия) и пожелтение кожи (желтуха). Блокированное кровообращение также может вызвать серьезное повреждение органов, включая инсульт. Заболевание имеет несколько признанных форм, включая серповидноклеточную анемию, серповидноклеточную болезнь гемоглобина C и серповидноклеточную бета-талассемию.
Признаки и симптомы
Гемоглобин — богатый железом белок, содержащийся в красных кровяных тельцах, который отвечает за перенос кислорода из легких к остальному телу. При СКА симптомы возникают из-за нарушением строения белка гемоглобина в эритроцитах (красные кровяные тельца). Нарушение гемоглобина приводит к тому, что красные кровяные тельца приобретают серповидную форму, что вызывает серию событий, приводящих к появлению хрупких красных кровяных телец и блокированию кровотока.
Наиболее частые признаки и симптомы серповидноклеточной анемии связаны с низким уровнем эритроцитов и болью. Наиболее частым симптомом анемии является чувство усталости и слабости (утомляемость). Эпизоды боли могут возникать внезапно и обычно появляются в костях и в брюшной полости, но могут возникать практически где угодно. Приступы боли длятся от нескольких дней до недели (острая) или продолжаются длительно (хроническая боль). Повреждение может произойти в большинстве частей тела, включая мозг, легкие, почки и суставы. СКА может вызвать пожелтение склеры глаз и кожи (желтуху) из-за распада крови. Признаки у младенцев могут включать опухшие и болезненные руки и/или ноги (дактилит), раздражительность, плач и тяжелые инфекции.
У пациентов с СКА может увеличиваться селезенка (спленомегалия), поскольку она улавливает эритроциты, которые присутствуют в кровотоке. Селезенка фильтрует аномальные красные кровяные тельца и борется с некоторыми инфекциями, такими как бактерии, вызывающие стрептококковое воспаление горла. Повреждение селезенки приводит к снижению способности бороться с некоторыми типично легкими инфекциями, которые могут стать опасными для жизни при СКА.
Если инфекция или серповидные клетки повреждают легкие может возникнуть острый грудной синдром. Это опасное для жизни осложнение СКА. Иногда у больных отсутствуют симптомы острого грудного синдрома, но в большинстве случаев больные испытывают боль в груди, затрудненное дыхание или жар. Дополнительные осложнения СКА включают инсульт, который может возникнуть у детей в возрасте от 2 лет. Мальчики и мужчины с серповидноклеточной анемией могут испытывать длительную болезненную эрекцию (приапизм) в любом возрасте.
Признаки и симптомы СКА варьируются от пациента к пациенту, и у некоторых пациентов симптомы более легкие, а у других могут быть более серьезные симптомы, требующие госпитализации. СКА присутствует при рождении; однако у большинства младенцев не появляются какие-либо признаки до четырехмесячного возраста, а у многих симптомы не проявляются до нескольких лет. Симптомы обычно появляются в первые три года жизни. Иногда первым признаком СКА является болезненный отек рук или ног младенца (дактилит). Эпизоды сильной боли часто вызываются чем-то вроде простуды, обезвоживания, инфекции, переутомления или травмы. Дети с СКА могут медленно расти и позже достичь половой зрелости.
С возрастом у пациентов с СКА все чаще возникают и другие дополнительные осложнения. Легочная гипертензия может развиться из-за повреждения мелких кровеносных сосудов и воздушных мешков в легких. Она приводит к снижению способности к физическим нагрузкам, одышку и усталость. Могут образоваться язвы на ногах, которые часто трудно заживают; повреждение сетчатки может вызвать проблемы с глазами. Повреждение суставов (аваскулярный некроз) и потеря костной ткани могут вызывать боль в суставах при ходьбе, стоянии и/или подъеме. Может произойти повреждение почек и часто присутствуют камни в желчном пузыре.
Есть много форм СКА. Наиболее распространенной тяжелой формой является S/S, которую просто называют серповидноклеточной анемией. Некоторые формы, такие как серповидная бета-талассемия, столь же серьезны, как и форма S/S. Серповидная бета-талассемия и серповидноклеточная болезнь гемоглобина С обычно менее тяжелы. Важно точно определить, какая форма СКА у пациента, и существует много путаницы в отношении различных форм.
Причины
Серповидноклеточная анемия вызывается мутациями в гене бета-гемоглобине (HBB) и наследуется по аутосомно-рецессивному типу. Генетические заболевания определяются двумя генами: один отца, а другой матери. Рецессивные генетические расстройства возникают, когда человек наследует один и тот же измененный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно бессимптомным. Риск для двух родителей-носителей передать измененный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем болезни, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%.
При СКА все сложнее. Люди, получившие серповидную мутацию от каждого родителя (S/S), страдают этим заболеванием. Некоторые люди с СКА получают мутацию только от одного родителя, но имеют мутацию, отличительную от мутации другого родителя. Это может привести к таким формам СКА, как серповидная бета-талассемия и серповидноклеточная болезнь гемоглобина C.
Говорят, что люди, унаследовавшие только одну мутацию гена HBB, имеют «серповидноклеточный признак». Эти люди, как правило, бессимптомные носители, которые могут передать ген своему потомству. У некоторых людей с серповидноклеточной патологией могут быть некоторые медицинские осложнения и проявляться некоторые симптомы.
Затронутые группы населения
Серповидноклеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причём больные серповидноклеточной анемией обладают повышенной (хотя и не абсолютной) врождённой устойчивостью к заражению различными штаммами малярийного плазмодия. СКА поражает 0,6% афроамериканского населения США (примерно 100 000 случаев в США). Болезнь также распространена у людей латиноамериканского происхождения из Индии, Центральной Америки и Аравийского полуострова, но может встречаться у людей любого происхождения. СКА поражает примерно одного из каждых 300–500 новорожденных афроамериканцев. Серповидноклеточная анемия присутствует примерно у 40 процентов населения в некоторых районах Африки. Частота серповидноклеточной анемии у американцев африканского происхождения составляет 9 процентов.
Мутации в гене HBB распространены у людей африканского, средиземноморского, ближневосточного и индийского происхождения, а также у людей из Карибского бассейна и некоторых регионов Центральной и Южной Америки, но их можно найти у людей любой национальности.
Диагностика
Врачи расценивают анемию, боли в желудке, костях и тошноту у чернокожих молодого возраста как возможные признаки серповидноклеточного криза. При подозрении на серповидноклеточную анемию они проводят анализы крови. Серповидные эритроциты и фрагменты разрушенных эритроцитов можно обнаружить при обследовании образца крови под микроскопом.
Также выполняется еще один анализ крови, который называют электрофорезом гемоглобина. При проведении электрофореза электрический ток используется для разделения различных типов гемоглобина, позволяющего обнаружить аномальный гемоглобин.
В зависимости от конкретных симптомов, возникающих у человека в период криза, может проводиться дополнительное тестирование. К примеру, если у человека наблюдается одышка или повышение температуры, может назначаться рентген грудной клетки.
Также доступно молекулярно-генетическое тестирование на мутации гена HBB.
Стандартные методы лечения
Профилактика — лучшее лечение. Информирование пациентов и их семей, лекарства, такие как гидроксикарбамид, предотвращение триггеров, раннее вмешательство и обследования для раннего выявления развивающихся осложнений, чтобы их можно было вылечить до того, как они станут серьезными, могут значительно улучшить результаты лечения.
Хотя очень немногие пациенты с СКА имеют возможность вылечиться, раннее направление новорожденных в специализированные центры может дать образование и ресурсы, которые помогут контролировать симптомы и значительно улучшить качество жизни пациентов. Больные с СКА должны проходить регулярные медицинские осмотры, где обучение предотвращению осложнений имеет огромную пользу для здоровья. Обучение семей тому, как тщательно контролировать детей на предмет лихорадки, находясь дома, давать низкие дозы пенициллина и иммунизировать, а также гарантировать, что семьи имеют информацию и возможность добраться до больницы, когда они заболеют, резко снижает тяжелые инфекции и смерть.
Для поддержания здоровья и сведения к минимуму боли и других осложнений можно выполнять множество простых действий, связанных с образом жизни. К ним относятся:
- сохранение гидратации;
- отсутствие чрезмерного тепла или холода;
- выполнение упражнений;
- глубокое дыхание;
- предотвращение утомляемости;
- избегание травм.
Как упоминалось выше, важно избегать болей. Как только возникает боль, важно использовать множество подходов для лечения сильной боли при серповидноклеточной анемии, а не только лекарства. Ключевым моментом является устранение триггеров боли, поэтому очень важны гидратация, сохранение тепла, прогулки и глубокое дыхание. Отвлечение может оказаться огромным подспорьем, как и такие подходы, как массаж, иглоукалывание и гипноз. Во время приступов боли можно вводить обезболивающие, такие как нестероидные противовоспалительные средства и опиатные анальгетики.
Переливание крови может быть использовано по многим причинам, таким как очень тяжелая анемия, подготовка к операции, а также для снижения риска или лечения инсульта. Некоторым пациентам может потребоваться хирургическое вмешательство из-за повреждения определенных органов, например, хирургия желчного пузыря (холецистэктомия) для удаления желчных камней.
Трансплантация стволовых клеток может излечить пациентов, но шансы на успех и потенциальные риски зависят от многих факторов.
Гидроксикарбамид (гидроксимочевина) был одобрен управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для лечения серповидноклеточной анемии и рекомендуется для большинства пациентов с S/S и серповидным бета-нулевой формами талассемии. Его следует предлагать детям с этими формами до 9 месяцев. Гидроксимочевина помогает стимулировать организм вырабатывать гемоглобин плода, тип гемоглобина, который есть у новорожденных, и снижает количество лейкоцитов, способствующих замедлению кровотока. В результате гидроксикарбамид может уменьшить боль, улучшить анемию, уменьшить количество госпитализаций и проблем с легкими, а также увеличить продолжительность жизни.
В 2017 году FDA одобрило препарат Эндари (L-глутамин) для пациентов в возрасте 5 лет и старше с СКА, чтобы уменьшить серьезные осложнения, связанные с заболеванием.
В 2019 году FDA одобрило два препарата для лечения СКА. Оксбрита (вокселотор) был одобрен для лечения СКА у взрослых и детей в возрасте 12 лет и старше, а Адаквео (кризанлизумаб) был одобрен в качестве лечения для снижения частоты вазоокклюзионных кризов у пациентов с СКА в возрасте 16 лет и старше.
Фолиевая кислота используется для обеспечения того, чтобы организм мог вырабатывать достаточно красных кровяных телец.
Пациентам и их семьям рекомендуется генетическое консультирование.
Прогноз
Продолжительность жизни людей с серповидноклеточной анемией сокращается. Однако некоторые пациенты могут оставаться без симптомов в течение многих лет, в то время как другие не выживают в младенчестве или раннем детстве. Тем не менее, при оптимальном лечении пациенты теперь могут выжить после четвертого десятилетия.
Большинство пациентов страдают периодическими болевыми кризами, усталостью, бактериальными инфекциями и прогрессирующими повреждениями тканей и органов. Нарушение роста и развития является результатом физических и эмоциональных травм, которые испытывают дети с серповидноклеточной анемией.
Причины смерти включают бактериальную инфекцию (наиболее частую причину), инсульт или кровоизлияние в мозг, а также почечную, сердечную или печеночную недостаточность. Риск бактериальных инфекций снижается после трехлетнего возраста. Тем не менее бактериальные инфекции — самая частая причина смерти в любом возрасте. Поэтому любые признаки инфекции у человека с серповидноклеточной анемией должны быть рассмотрены врачом, чтобы предотвратить повреждение и спасти жизни.