From Wikipedia, the free encyclopedia
In vascular diseases, endothelial dysfunction is a systemic pathological state of the endothelium. Along with acting as a semi-permeable membrane, the endothelium is responsible for maintaining vascular tone and regulating oxidative stress by releasing mediators, such as nitric oxide, prostacyclin and endothelin, and controlling local angiotensin-II activity.[1][2]
Research[edit]
Atherosclerosis[edit]
Endothelial dysfunction may be involved in the development of atherosclerosis[3][4][5] and may predate vascular pathology.[3][6] Endothelial dysfunction can also lead to increased adherence of monocytes and macrophages, as well as promoting infiltration of LDL in the vessel wall.[7] Dyslipidemia and hypertension are well known to contribute to endothelial dysfunction,[8][9] and lowering blood pressure and LDL has been shown to improve endothelial function, particularly when lowered with ACE inhibitors, calcium channel blockers, and statins.[10]
Nitric oxide[edit]
Nitric oxide (NO) suppresses platelet aggregation, inflammation, oxidative stress, vascular smooth muscle cell migration and proliferation, and leukocyte adhesion.[4] A feature of endothelial dysfunction is the inability of arteries and arterioles to dilate fully in response to an appropriate stimulus, such as exogenous nitroglycerine,[3] that stimulates release of vasodilators from the endothelium like NO. Endothelial dysfunction is commonly associated with decreased NO bioavailability, which is due to impaired NO production by the endothelium or inactivation of NO by reactive oxygen species.[citation needed]
Testing and diagnosis[edit]
In the coronary circulation, angiography of coronary artery responses to vasoactive agents may be used to test for endothelial function, and venous occlusion plethysmography and ultrasonography are used to assess endothelial function of peripheral vessels in humans.[3]
A non-invasive method to measure endothelial dysfunction is % Flow-Mediated Dilation (FMD) as measured by Brachial Artery Ultrasound Imaging (BAUI).[11] Current measurements of endothelial function via FMD vary due to technical and physiological factors. Furthermore, a negative correlation between percent flow mediated dilation and baseline artery size is recognised as a fundamental scaling problem, leading to biased estimates of endothelial function.[12]
A non-invasive, FDA-approved device for measuring endothelial function that works by measuring Reactive Hyperemia Index (RHI) is Itamar Medical’s EndoPAT.[13][14] It has shown an 80% sensitivity and 86% specificity to diagnose coronary artery disease when compared against the gold standard, acetylcholine angiogram.[15] This results suggests that this peripheral test reflects the physiology of the coronary endothelium.
Since NO maintains low tone and high compliance of the small arteries at rest,[16] a reduction of age-dependent small artery compliance is a marker for endothelial dysfunction that is associated with both functional and structural changes in the microcirculation.[17] Small artery compliance or stiffness can be assessed simply and at rest and can be distinguished from large artery stiffness by use of pulsewave analysis.[18]
Endothelial dysfunction and stents[edit]
Stent implantation has been correlated with impaired endothelial function in several studies.[19] Sirolimus eluting stents were previously used because they showed low rates of in-stent restenosis, but further investigation showed that they often impair endothelial function in humans and worsen conditions.[19] One drug used to inhibit restenosis is iopromide-paclitaxel.[20]
Risk reduction[edit]
Treatment of hypertension and hypercholesterolemia may improve endothelial function in people taking statins (HMGCoA-reductase inhibitor), and renin angiotensin system inhibitors, such as ACE inhibitors and angiotensin II receptor antagonists.[21][22] Calcium channel blockers and selective beta 1 antagonists may also improve endothelial dysfunction.[10] Life style modifications such as smoking cessation have also been shown to improve endothelial function and lower the risk of major cardiovascular events.[23]
See also[edit]
- Atherosclerosis
- Endothelial activation
- Nitric oxide
- endothelial nitric oxide synthase
- Prostacyclin
- Endothelium-derived relaxing factor
- Endothelin
- Integrin network
- Endothelial shear stress
References[edit]
- ^ Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P. (2010). «From endothelial dysfunction to atherosclerosis». Autoimmunity Reviews. 9 (12): 830–834. doi:10.1016/j.autrev.2010.07.016. PMID 20678595.
- ^ Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, Hamburg NM, Lüscher TF, Shechter M, Taddei S, Vita JA, Lerman A (Aug 2012). «The assessment of endothelial function: from research into clinical practice». Circulation. 126 (6): 753–67. doi:10.1161/circulationaha.112.093245. PMC 3427943. PMID 22869857.
- ^ a b c d Maruhashi, T; Kihara, Y; Higashi, Y (2018). «Assessment of endothelium-independent vasodilation: From methodology to clinical perspectives». Journal of Hypertension. 36 (7): 1460–1467. doi:10.1097/HJH.0000000000001750. PMID 29664811. S2CID 4948849.
- ^ a b Eren E, Yilmaz N, Aydin O (2013). «Functionally defective high-density lipoprotein and paraoxonase: a couple for endothelial dysfunction in atherosclerosis». Cholesterol. 2013: 792090. doi:10.1155/2013/792090. PMC 3814057. PMID 24222847.
- ^ Botts SR, Fish JE, Howe KL (December 2021). «Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment». Frontiers in Pharmacology. 12: 787541. doi:10.3389/fphar.2021.787541. PMC 8727904. PMID 35002720.
- ^ Münzel T, Sinning C, Post F, Warnholtz A, Schulz E (2008). «Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction». Annals of Medicine. 40 (3): 180–96. doi:10.1080/07853890701854702. PMID 18382884. S2CID 18542183.
- ^ Poredos, P. (2001). «Endothelial dysfunction in the pathogenesis of atherosclerosis». Clinical and Applied Thrombosis/Hemostasis. 7 (4): 276–280. doi:10.1177/107602960100700404. ISSN 1076-0296. PMID 11697708. S2CID 71334997.
- ^ Le Master, Elizabeth; Levitan, Irena (2019-01-22). «Endothelial stiffening in dyslipidemia». Aging. 11 (2): 299–300. doi:10.18632/aging.101778. ISSN 1945-4589. PMC 6366977. PMID 30674709.
- ^ Konukoglu, Dildar; Uzun, Hafize (2017). «Endothelial Dysfunction and Hypertension». Advances in Experimental Medicine and Biology. 956: 511–540. doi:10.1007/5584_2016_90. ISBN 978-3-319-44250-1. ISSN 0065-2598. PMID 28035582.
- ^ a b Ghiadoni, Lorenzo; Taddei, Stefano; Virdis, Agostino (2012). «Hypertension and endothelial dysfunction: therapeutic approach». Current Vascular Pharmacology. 10 (1): 42–60. doi:10.2174/157016112798829823. ISSN 1875-6212. PMID 22112351.
- ^ Peretz, Alon; Daniel F Leotta; Jeffrey H Sullivan; Carol A Trenga; Fiona N Sands; Mary R Aulet (2007). «Flow mediated dilation of the brachial artery: an investigation of methods requiring further standardization». BMC Cardiovascular Disorders. 7 (11): 11. doi:10.1186/1471-2261-7-11. PMC 1847451. PMID 17376239.
- ^ Thijssen DH, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, Parker B, Widlansky ME, Tschakovsky ME, Green DJ (Jan 2011). «Assessment of flow-mediated dilation in humans: a methodological and physiological guideline». Am J Physiol Heart Circ Physiol. 300 (1): H2–12. doi:10.1152/ajpheart.00471.2010. PMC 3023245. PMID 20952670.
- ^ Kuvin JT, Mammen A, Mooney P, Alsheikh-Ali AA, Karas RH (Feb 2007). «Assessment of peripheral vascular endothelial function in the ambulatory setting». Vasc. Med. 12 (1): 13–6. doi:10.1177/1358863×06076227. PMID 17451088.
- ^ Axtell AL, Gomari FA, Cooke JP (October 2010). «Assessing endothelial vasodilator function with the Endo-PAT 2000». Journal of Visualized Experiments (44). doi:10.3791/2167. PMC 3143035. PMID 20972417.
- ^ Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Kuvin JT, Lerman A (Dec 2004). «Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia». J Am Coll Cardiol. 44 (11): 2137–41. doi:10.1016/j.jacc.2004.08.062. PMID 15582310.
- ^ Gilani M, Kaiser DR, Bratteli CW, Alinder C, Rajala Bank AJ, Cohn JN (2007). «Role of nitric oxide deficiency and its detection as a risk factor in pre-hypertension». JASH. 1 (1): 45–56. doi:10.1016/j.jash.2006.11.002. PMID 20409832.
- ^ Duprez DA, Jacobs DR, Lutsey PL, Bluemke FA, Brumback LC, Polak JF, Peralta CA, Greenland P, Kronmal RA (2011). «Association of small artery elasticity with incident cardiovascular disease in older adults: the multiethnic study of atherosclerosis». Am J Epidemiol. 174 (5): 528–36. doi:10.1093/aje/kwr120. PMC 3202150. PMID 21709134.
- ^ Cohn JN, Duprez DA, Finkelstein SM (2009). «Comprehensive noninvasive arterial vascular evaluation». Future Cardiology. 5 (6): 573–9. doi:10.2217/fca.09.44. PMID 19886784.
- ^ a b Bedair, T. M; Elnaggar, M. A; Joung, Y. K; Han, D. K (2017). «Recent advances to accelerate re-endothelialization for vascular stents». Journal of Tissue Engineering. 8: 2041731417731546. doi:10.1177/2041731417731546. PMC 5624345. PMID 28989698.
- ^ Unverdorben, Martin; Vallbracht, Christian; Cremers, Bodo; Heuer, Hubertus; Hengstenberg, Christian; Maikowski, Christian; Werner, Gerald S.; Antoni, Diethmar; Kleber, Franz X. (2009-06-16). «Paclitaxel-coated balloon catheter versus paclitaxel-coated stent for the treatment of coronary in-stent restenosis». Circulation. 119 (23): 2986–2994. doi:10.1161/circulationaha.108.839282. ISSN 0009-7322. PMID 19487593.
- ^ Ruilope LM, Redón J, Schmieder R (2007). «Cardiovascular risk reduction by reversing endothelial dysfunction: ARBs, ACE inhibitors, or both? Expectations from the ONTARGET Trial Programme». Vascular Health and Risk Management. 3 (1): 1–9. PMC 1994043. PMID 17583170.
- ^ Briasoulis A, Tousoulis D, Androulakis ES, Papageorgiou N, Latsios G, Stefanadis C (Apr 2012). «Endothelial dysfunction and atherosclerosis: focus on novel therapeutic approaches». Recent Pat Cardiovasc Drug Discov. 7 (1): 21–32. doi:10.2174/157489012799362386. PMID 22280336.
- ^ Messner, Barbara; Bernhard, David (2014). «Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis». Arteriosclerosis, Thrombosis, and Vascular Biology. 34 (3): 509–515. doi:10.1161/ATVBAHA.113.300156. ISSN 1524-4636. PMID 24554606.
Клиническое значение эндотелиальной дисфункции в хирургии
Статьи
РОССИЙСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
КАФЕДРА ФАКУЛЬТЕТСКОЙ ХИРУРГИИ ИМ. С.И. СПАСОКУКОЦКОГО
ПЕРВАЯ ГРАДСКАЯ БОЛЬНИЦА ИМ. Н.И. ПИРОГОВА
В.С. Савельев, В.А. Петухов
АКТУАЛЬНОСТЬ
ЭНДОТЕЛИЙ — активный эндокринный орган, дисфункция которого является обязательным компонентом патогенеза всех сердечно-сосудистых заболеваний, воспалительных и аутоиммунных реакций, диабета, тромбоза, сепсиса, роста злокачественных опухолей. Масса этого органа составляет около 1,5 — 1,7 кг; длина монослоя эндотелия приближается к 7 км, а площадь поверхности эндотелия равна площади футбольного поля.
ФУНКЦИИ ЭНДОТЕЛИЯ
Эндотелий обеспечивает гомеостаз путём регуляции равновесного состояния противоположных процессов:
ЭНДОТЕЛИАЛЬНАЯ ДИСФУНКЦИЯ
Определение: неадекватное (увеличенное или сниженное) образование в эндотелии различных биологических веществ.
Синоним: патологическое состояние эндотелия, возникающее при нарушении продукции оксида азота.
КЛИНИЧЕСКИЕ АСПЕКТЫ:
РЕАЛЬНАЯ ПРИЧИНА ДИСФУНКЦИИ ЭНДОТЕЛИЯ — ХРОНИЧЕСКАЯ ЭНДОТОКСИНОВАЯ АГРЕССИЯ
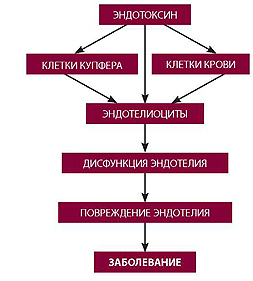
Схема 1. Эндотелиальная дисфункция (этиопатогенез)
СТАДИИ ПОВРЕЖДЕНИЯ ЭНДОТЕЛИЯ
I. ПОВРЕЖДЕНИЕ ГЛИКОКАЛИКСА ЭНДОТЕЛИЯ
Эндотелиальный гликокаликс:
II. УВЕЛИЧЕНИЕ ПРОНИЦАЕМОСТИ ЭНДОТЕЛИЯ
Патогенез атеросклероза и воспалительных заболеваний всегда следует рассматривать через призму нарушений эндотелиального цитоскеле-та, т.е., проницаемости.
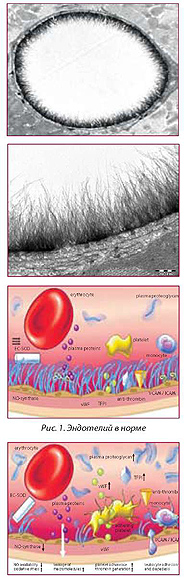
Рис. 2. Эндотоксиновая агрессия
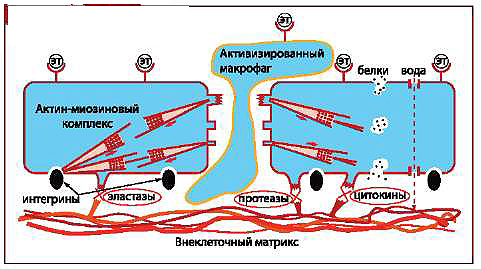
Рис. 3. Активация киназы легких цепей миозина и увеличение проницаемости эндотелия
III. ПОВРЕЖДЕНИЕ ЭНДОТЕЛИЯ — АПОПТОЗ И АНОИКОЗ ЭНДОТЕЛИОЦИТОВ
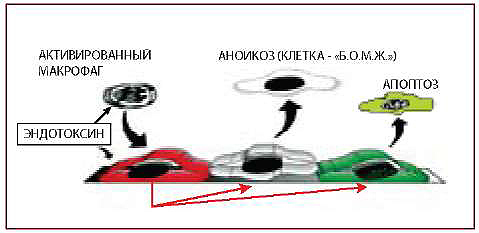
Рис. 4. Аноикоз и апоптоз образуют «бреши» в эндотелии (тромбоз, атеросклероз и пр.)
АПОПТОЗ (APOPTOSIS)
АНОИКОЗ (ANOIKIS)
ДИАГНОСТИКА ДИСФУНКЦИИ ЭНДОТЕЛИЯ
< 1 мг/мл — минимальный; 1,1-1,9 мг/мл — низкий; 2,0-2,9 мг/мл — средний;
> 3 мг/мл — высокий
ЛЕЧЕНИЕ
Существующие методы лечения дисфункции эндотелия («заместительная терапия», влияние на синтез эндотелиальных факторов, уменьшение связывания эндотелия с прокоагулянтами, гиполипидемическая терапия, влияние на экспрессию молекул адгезии и влияние на апоптоз эндотелиоцитов) применяются в комплексе либо раздельно, не имеют этиопатогенетичес-кой базы, их следует относить к различным вариантам симптоматической терапии.
ЭТИОПАТОГЕНЕТИЧЕСКИЙ ПОДХОД К ЛЕЧЕНИЮ ЭНДОТЕЛИАЛЬНОЙ ДИСФУНКЦИИ:
I. ЭНДОТОКСИНСВЯЗЫВАЮЩИЙ КОМПЛЕКС:
II. ЭНДОТЕЛИОПРОТЕКЦИЯ: блокада или уменьшение синтеза фермента киназы лёгких цепей миозина эндотелиоцитов изокверцетином и кверцетин-глюкуронидом красных листьев винограда (растительный эндотелиопротектор АНТИСТАКС® ).
ДИЗАЙН ИССЛЕДОВАНИЯ
Все пациенты перенесли распространённый перитонит.
Лечебная группа:
Контрольная группа:
Этапы исследования:
1) при выписке из стационара;
2) через 4 месяца непрерывной терапии. Лечебная и контрольная группы сопоставимы по полу, возрасту, срокам после операции, сопутствующим заболеваниям.
ВЫВОДЫ:
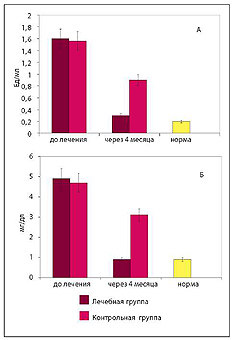
Рис. 5. Концентрация эндотоксина (А) и высокочувствительного СРБ (Б) в плазме крови
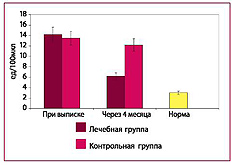
Рис. 6. Количество циркулирующих эндотелиоцитов в плазме крови
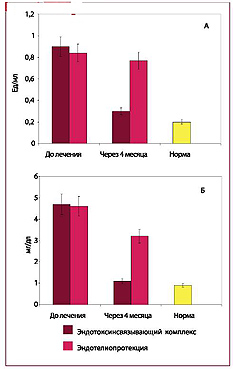
Рис. 7. Концентрации эндотоксина (А) и высокочувствительного СРБ (Б) в плазме крови
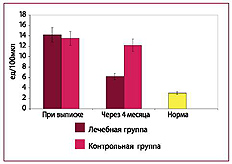
Рис. 8. Количество циркулирующих эндотелиоцитов в плазме крови
ВОПРОСЫ И ОТВЕТЫ
ВОПРОС 1: Какова эффективность этиопатоге-нетической терапии эндотелиальной дисфункции?
ОТВЕТ: Комплексная терапия приводит к достоверному снижению концентрации эндотоксина и СРБ, а также позволяет уменьшить повреждение эндотелия.
ВОПРОС 2: Играют ли изокверцетин и кверце-тин- глюкуронид красных листьев винограда роль эндотелиопротекторов в комплексной терапии дисфункции эндотелия или нужно ли пациентам назначать АНТИСТАКС®?
ОТВЕТ: Эндотелиопротекторы не снижают эндо-токсинемию и не влияют на концентрацию СРБ в крови. Эндотоксинсвязывающий комплекс не предотвращает повреждение эндотелиоцитов, для этого необходима эндотелиопротекция изок-верцетином и кверцетином- глюкуронидом.
ПРОФИЛАКТИКА И ЛЕЧЕНИЕ ЭНДОТЕЛИ АЛЬНОЙ ДИСФУНКЦИИ В ЭКСТРЕННОЙ ХИРУРГИИ (после операции)
1. ЭНТЕРОСОРБЦИЯ ФИШант-С® 1 раз в неделю по 200 г (4 — 12 месяцев);
2. ЭНДОТЕЛИОПРОТЕКЦИЯ растительным эндотелиопротектором АНТИСТАКС® (по 4 капсулы в сутки 1 месяц, далее по 2 капсулы — 3 месяца);
3. ЛЕЧЕНИЕ нарушений метаболических функций печени растительными комбинированными гепатопротектором ГЕПАБЕНЕ® (по 2 капсулы 3 раза в сутки 8 — 10 недель);
4. ВОССТАНОВЛЕНИЕ транспорта желчи в 12-ти перстную кишку растительным избирательным спазмолитическим средством БУСКО-ПАН® (по 20 мг 2 раза в сутки 2 — 3 месяца);
5. ВОССТАНОВЛЕНИЕ микробиоценоза толстой кишки пребиотиками метаболического типа ХИЛАК ФОРТЕ® (по 20 — 40 капель 3 раза в сутки 8 — 16 недель).
ПРОФИЛАКТИКА И ЛЕЧЕНИЕ ЭНДОТЕ-ЛИАЛЬНОЙ ДИСФУНКЦИИ В ПЛАНОВОЙ ХИРУРГИИ
(предоперационная подготовка, после операции)
I. ХИРУРГИЧЕСКИЕ ВМЕШАТЕЛЬСТВА БЕЗ АНТИБАКТЕРИАЛЬНОЙ ПРОФИЛАКТИКИ:
А. ПРЕДОПЕРАЦИОННАЯ подготовка в период амбулаторного обследования:
Б. ПОСЛЕОПЕРАЦИОННАЯ профилактика:
II. АНТИБИОТИКОСОПРОВОЖДАЕМЫЕ ХИРУРГИЧЕСКИЕ ВМЕШАТЕЛЬСТВА
(послеоперационное лечение):
ДЛИТЕЛЬНОСТЬ ЛЕЧЕНИЯ ПРИ АНТИБИ-ОТИКОСОПРОВОЖДАЕМЫХ ХИРУРГИЧЕСКИХ ВМЕШАТЕЛЬСТВАХ
Количество суток антибактериальной терапии = количеству недель этиопатогенетической терапии эндотелиальной дисфункции.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)

Введение
Эндотелий сосудов играет важную роль в регуляции сосудистого тонуса в норме и при различных заболеваниях. Под термином «функция эндотелия» принято подразумевать регуляцию капиллярного кровотока, осуществляемую за счет динамической смены фаз вазоконстрикции и вазодилатации сосудов резистивного типа в соответствии с потребностями клеточного обмена веществ [1, 2], соответственно, «дисфункция эндотелия» — это нарушение регуляции динамической реакции сосудов в ответ на соответствующие раздражители. Дисфункция эндотелия лежит в основе множества патологических состояний, таких как атеросклероз, артериальная гипертензия, преэклампсия [1–3].
Эндотелиальные клетки (ЭК) являются клетками внутренней оболочки сосудов и играют важную роль в процессе тканевого дыхания и метаболизма. Нормальные ЭК взрослого человека остаются в основном неподвижными, но могут быстро активироваться в ответ на травму или патологические состояния, когда требуется ангиогенез [4]. Ангиогенез регулируется тремя основными подтипами ЭК, которые выполняют специализированные задачи: клетки, инициирующие ангиогенез, которые направляют рост сосудистого отростка в ответ на факторы роста; стеблевые клетки, которые разрастаются и удлиняют росток; покоящиеся клетки, которые присутствуют в новообразующихся сосудах и регулируют сосудистый гомеостаз и функцию эндотелиального барьера [5, 6]. В обзоре представлены данные литературы о функции и дисфункции ЭК. Нами проведен поиск и анализ опубликованных полнотекстовых обзорных и оригинальных статей на иностранном (английском) и русском языках с использованием баз данных eLIBRARY.RU, Google Scholar, Web of Science, Scopus и PubMed за период с 2004 по 2021 г. Приоритет отдавался оригинальным статьям, посвященным исследованиям состояния эндотелия, а также его изменениям при различных заболеваниях у людей. Для поиска были использованы следующие ключевые слова: эндотелий, дисфункция эндотелия, физиология эндотелия.
Процесс неоваскулогенеза
Сосудистая сеть раньше всех остальных органов формируется в процессе онтогенеза и впоследствии созревает в замкнутую сложную систему сосудов различного диаметра. Все органы и ткани организма, за исключением хрящевой ткани и роговицы, зависят от тока крови, необходимого для осуществления процессов жизнедеятельности [4, 7].
Процесс васкулогенеза начинается на раннем этапе развития эмбриона. Мезодермальные ангиобласты объединяются с образованием примитивных сосудистоподобных трубок, лишенных стенки, также в процессе первичного ангиогенеза принимают участие гемангиобласты, впоследствии дифференцирующиеся в эндотелиальные и гемопоэтические клетки [8, 9].
Последующее ремоделирование сосудистого русла достигается двумя механизмами: инвагинацией и прорастанием сосудов. Инвагинация приводит к расширению капиллярного русла за счет «разделения» капилляра на два соседних сосуда, при этом противоположные стенки первичного сосуда выступают в его просвет, происходит контакт ЭК друг с другом, чтобы сформировать локальный эндотелиальный бислой, с имеющимися связями между ЭК. Перициты и миофибробласты покрывают образовавшийся полый транскапиллярный столб, который увеличивается по окружности, разделяя капилляр на два параллельных сосуда [10].
Прорастание сосудов возникает в результате увеличивающейся потребности тканей в кислороде, что стимулирует выработку факторов роста эндотелия сосудов (vascular endothelial growth factor, VEGF), факторов роста фибробластов и других проангиогенных факторов. VEGF стимулируют рецепторы на поверхности эндотелия, в результате чего развивается локальная релаксация сосуда, происходит разрушение контактов между эндотелиоцитами, отделение перицитов и разрушение базальной мембраны. Далее происходит миграция клеток эндотелия и удлинение будущего сосудистого ростка. При этом дифференцируются концевые и стеблевые ЭК [11].
Несмотря на то, что процесс прорастания происходит из ЭК одного и того же сосуда, концевые и стеблевые клетки в формирующемся сосуде различаются как функционально, так и морфологически. Концевые клетки имеют многочисленные филоподии и выступы, соответствующие их высокоподвижному поведению, тогда как у стеблевых клеток относительно мало филоподий [12].
Интересно, что ЭК, являющаяся клеткой-инициатором прорастания, «навязывает» фенотип клеткам посредством экспрессии лиганда Notch Delta-like 4 (Dll4). В соседних ЭК Dll4 связывает рецепторы Notch, вызывая высвобождение внутриклеточного домена Notch и управление экспрессией рецептора VEGF1 (VEGFR1) [13], на фоне снижения экспрессии VEGFR2. Повышенное соотношение VEGFR1/VEGFR2 снижает чувствительность ЭК к VEGF и «навязывает» фенотип стеблевых клеток [14].
Особенности метаболизма эндотелиальных клеток
В метаболизме ЭК основную роль занимает процесс гликолиза, который имеет ряд преимуществ перед окислительным фосфорилированием: во-первых, высокая скорость гликолиза поддерживает продукцию лактата, который функционирует как проангиогенная сигнальная молекула [15, 16]. Во-вторых, активные формы кислорода сохраняются на минимальном уровне, тогда как количество кислорода, доступного для передачи тканям, остается на достаточном уровне [17]; в-третьих, зависимость от гликолиза создает предпосылки для прорастания ЭК в бессосудистую, гипоксическую среду, где уровни интерстициальной глюкозы не ограничивают скорость процесса [18, 19].
Активность процесса гликолиза напрямую зависит от стимуляции VEGF, которые способны повышать уровень экспрессии переносчика глюкозы 1 и гликолитических ферментов, таких как лактатдегидрогеназа (ЛДГ) A и бифункциональная 6-фосфофрукто-2-киназа/фруктозо-2,6-бисфосфатаза-3 (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3, PFKFB3). Последняя является регулятором гликолиза и использует свою киназную активность (которая в 700 раз превышает фосфатазную активность) для выработки фруктозо-2,6-бисфосфата, который аллостерически активирует ограничивающий скорость гликолитический фермент фосфофруктокиназу-1. Несмотря на то, что генетически обусловленный дефицит или химическое ингибирование PFKFB3 лишь частично (на 40%) снижает гликолиз, этого достаточно, чтобы существенно нарушить прорастание ЭК in vitro, а также ветвление и разрастание сосудов in vivo [20–22]. В зрелом эндотелии наблюдается снижение активности гликолиза и уменьшение количества митохондрий, что обусловливает функциональный покой эндотелия [23].
Следует отметить, что количество митохондрий в эндотелии составляет примерно 2–6%, при этом в гепатоцитах их содержится 28%. Однако при переходе из состояния покоя в ангиогенез потребление кислорода в ЭК усиливается в 3 раза [24]. При этом работа митохондрий эндотелия согласуется с эффектом Крэбтри, при котором более низкие уровни глюкозы (~1 ммоль/л) вызывают усиление митохондриального дыхания с противоположными эффектами (ингибирование роста и снижение дыхания) при высоких уровнях глюкозы [25].
Особенности обмена липидов в эндотелиальных клетках
Эндотелиальные клетки не только способны накапливать липиды, но также самостоятельно их синтезировать. Поскольку ферменты синтеза триглицеридов находятся в эндоплазматическом ретикулуме, образование липидных капель de novo предположительно происходит в его мембране. При необходимости липиды гидролизируются с образованием жирных кислот при участии триглицеридлипазы жировой ткани, гормоночувствительной липазы и моноглицеридлипазы [26]. Кавеолины (Cav-1–3) представляют собой белки оболочки, управляющие биогенезом кавеол, т. е. микродоменов липидных рафтов с колбообразной структурой выпячивания 60–100 нм. Потеря эндотелиального Cav-1 нарушает образование липидных капель за счет усиленного липолиза под влиянием гормоночувствительной липазы, что, возможно, объясняет, почему мыши с дефицитом Cav-1 защищены от атеросклероза [27]. Образование липидных капель в ЭК необходимо для предотвращения липотоксичности, обеспечения процесса β-окисления жирных кислот для снижения интенсивности процесса гликолиза и высвобождения жирных кислот из ЭК в соседние периваскулярные клетки [26].
Таким образом, ЭК принимают активное участие в обмене липидов: синтез липидов в ЭК необходим для их миграции, ингибирование ацетил-КоА-карбоксилазы сдвигает липидный состав мембран ЭК в сторону увеличения уровня полиненасыщенных жирных кислот, что снижает текучесть мембран, образование филоподий и миграцию ЭК [28]. Наличие липидов в ЭК способно вызвать дисфункцию эндотелия: окисленные фосфолипиды, увеличивают секрецию пуринов, при этом для поддержания клеточного уровня АТФ ЭК увеличивают синтез глицина посредством регуляции митохондриальной метилентетрагидрофолат дегидрогеназы/циклогидролазы [29]. ЭК транспортируют липиды в другие клетки. При этом важное значение в этом процессе имеет транслоказа жирных кислот FAT/CD36, отвечающая за перенос жирных кислот через клеточную мембрану. Внутри ЭК липиды находятся либо в свободном состоянии в виде жирных кислот, либо связаны с белками, связывающими жирные кислоты, которые транспортируют жирные кислоты к местам назначения [30].
Таким образом, эндотелий сосудов играет жизненно важную и повсеместную роль в сосудистом гомеостазе, регулируя транспорт клеток, питательных веществ и метаболитов между кровотоком и подлежащими тканями. Сахарный диабет, ожирение, дислипидемия, курение способны вызывать дисфункцию эндотелия, проявлением которой могут быть: повреждение и утрата целостности с увеличением проницаемости сосудистой стенки, индукция синтеза цитокинов и молекул адгезии, метаболические нарушения, создание протромботической среды, дедифференциация клеток [31].
Функционирование эндотелия в условиях гипоксии
При тканевой гипоксии увеличивается экспрессия факторов, индуцируемых гипоксией (hypoxia-inducible factors, HIF-факторы), за счет пролилгидроксилазы (prolyl hydroxylase domain, PHD). PHD необходим кислород для гидроксилирования субъединицы HIFa. Во время гипоксии PHD теряет способность гидроксилировать HIF из-за их ферментативной зависимости от кислорода, и потеря этого механизма деградации приводит к активации HIF-опосредованной программы транскрипции, которая включает в себя индукцию ангиогенеза, метаболизма глюкозы и рассматривается как важный фактор в развитии злокачественных опухолей. HIF транскрипционно функционирует как гетеродимер, состоящий из субъединиц HIFa и HIFbs, который связывается с элементом ответа на гипоксию в промоторе генов-мишеней. В большинстве типов клеток HIF-1 экспрессируется при острой гипоксии. Переход от HIF-1 к HIF-2 наблюдается в случае хронизации процесса гипоксии, несмотря на то, что большинство генов регулируется и тем и другим фактором одновременно [32]. HIF-2α увеличивает экспрессию тирозинфосфатазы, что, в свою очередь, снижает фосфорилирование V-кадерина, поддерживая целостность связи, и предотвращает потерю барьерной функции эндотелия [33]. Экспрессия HIF-1α в альвеолярных ЭК усиливает реакцию воспаления и способствует клеточноопосредованному воспалению с активацией CD4+ и CD8+, а также увеличивает экспрессию провоспалительных цитокинов интерлейкина (ИЛ) 2 и фактора некроза опухоли-α, которые подавляют CD55, в результате чего происходит усиление комплемент-ассоциированного повреждения эндотелия [34]. Кроме того, HIF-1α миелоидных клеток является ключевым фактором активации клеток в условиях гипоксии и воспаления за счет модуляции клеточной энергетики, активации гликолитических ферментов и транспортеров глюкозы, что позволяет генерировать АТФ в условиях гипоксии и предотвращать апоптоз клеток врожденного иммунитета. Однако при хронических инфекциях HIF-1α предотвращает чрезмерное рекрутирование лимфоцитов в интерстиций легких и иммунопатологические последствия для организма хозяина [35]. Увеличение количества циркулирующих ЭК-предшественников положительно коррелирует с выживаемостью пациентов [36].
Повреждение эндотелия при COVID-19
Исходное повреждение эндотелия обнаруживается у пациентов с сахарным диабетом и ожирением за счет повышения содержания адипокинов в плазме: этот эффект связан с активацией на фоне воспаления криопирина и аутокринной продукцией ИЛ-1β [37]. Присоединение инфекции усиливает имеющееся повреждение эндотелия, что вызывает избыточное образование тромбина и снижение фибринолиза [38, 39]. Тромбин способствует дальнейшему повреждению эндотелия, которое можно предотвратить in vitro с помощью агонистов хемокинового (мотив С-Х-С) рецептора-4 (CXCR4), таких как убиквитин [40]. Более того, гипоксия может привести к увеличению экспрессии HIF-1α и гиперкоагуляции [41]. Таким образом, у пациентов с пневмонией COVID-19 регистрируется более высокая частота тромботических эпизодов, в то время как повышенная проницаемость сосудов, по-видимому, тесно связана с повышенным тромбозом. В частности, у пациентов с пневмонией и органной недостаточностью повышенная проницаемость сосудов сильно коррелировала с тяжелой лимфопенией [42].
При проведении КТ органов грудной клетки у пациентов с COVID-19 обнаружено более раннее появление интерстициального отека легких по сравнению с пациентами, имеющими острый респираторный дистресс-синдром (ОРДС), с последующим присоединением альвеолярного отека, что ставит под сомнение сходство повреждения легких при COVID-19 и ОРДС. При патогистологическом исследовании образцов легочной ткани, взятых у пациентов, умерших от COVID-19, обнаруживается диффузный микроциркуляторный и макрососудистый тромбоз, что не характерно для ОРДС [43]. При этом признаков васкулита и ДВС-синдрома не наблюдается: количество антитромбина-III, фибриногена и уровень тромбоцитов незначительно снижаются на ранних этапах заболевания, в то время как уровень D-димера прогрессивно увеличивается и является прогностическим признаком тяжелого течения COVID-19 [44].
Апоптоз клеток эндотелия сосудов легких может быть также вызван наличием хронического воспаления, например при хронической обструктивной болезни легких, или остро возникать на фоне ОРДС; в последнем случае он активируется киназой Брутона, ИЛ-17. На фоне повреждения эндотелия наблюдается выделение ЛДГ в кровь апоптозными ЭК [45]. Апоптоз клеток эндотелия также может возникать на фоне вирусных инфекций путем аутофагии, которая индуцируется НАДФН-оксидазой-2 [46]. Кроме того, эндотелий лимфатических сосудов легких наиболее чувствителен к окислительному стрессу, и при инфицировании SARS-CoV-2 именно эта популяция клеток подвергается наибольшему повреждению [47].
До настоящего времени остается спорным вопрос участия тромбоцитов в процессе повреждения эндотелия при COVID-19. Известно, что низкое количество тромбоцитов увеличивает в 5 раз смертность от COVID-19, хотя опубликованные показатели неоднородны. Чаще у пациентов с COVID-19 наблюдается увеличение уровня тромбоцитов, что, вероятно, связано с повышением содержания в сыворотке тромбопоэтина на фоне пневмонии [48–50].
Заключение
Пандемия COVID-19 заставила обратить более пристальное внимание на изучение свойств эндотелия и предоставить практическому здравоохранению инструменты для патогенетически обоснованной терапии заболеваний, связанных с патологией эндотелия. Представленный обзор данных литературы позволяет еще раз обозначить проблему эндотелиальной дисфункции, увидеть, что эндотелий является уникальной структурой, регулирующей деятельность всего макроорганизма, а нарушение функции ЭК является важным патогенетическим механизмом, лежащим в оcнове генеза различных заболеваний. Несмотря на то, что имеются сведения о маркерах дисфункции эндотелия, таких как HIF, VEGF, на наш взгляд, необходим дальнейший поиск новых маркеров, применимых в рутинной клинической практике. Безусловно важным направлением выступает поиск терапевтических стратегий коррекции эндотелиальной дисфункции.
1. Алмазов В. А., Беркович О. А., Ситникова М. Ю. и др. Эндотелиальная дисфункция у больных с дебютом ишемической болезни в разном возрасте // Кардиология. 2001. № 5. С. 26-29.
2. Бувальцев В.И., Машина С.Ю., Покидышев Д.А. и др. Роль коррекции метаболизма оксида азота в организме при профилактике гипертонического ремоделирования сердечно-сосудистой системы // Росс. кард. журн. 2002. № 5. С. 74-81.
3. Васина Л.В. Клеточные и гуморальные маркеры апоптоза при остром коронарном синдроме в сочетании с гипертонической болезнью // Артериальная гипертензия. 2008. Т.14. № 4. С. 332-335.
4. Дисфункция эндотелия. Патогенетическое значение и методы коррекции / Под ред. Н.Н. Петрищева. СПб.: НИЦВМА, 2007. — 296 с.
5. Князькова И.И., Цыганков А.М., Далашзаде С.Р. Влияние каптоприла на изменение эндотелиальных факторов у больных с острым инфарктом миокарда // Украинский кардиологический журнал. 2004. № 7. С. 34-38.
6. Комилова М.С., Пахомова Ж.Е. Значение эндотелия в развитии осложнений гестационного периода // Росс. вестн. акуш-гинекол. 2015. Т. 15. № 1. С. 18-23.
7. Крукиер И.И. Плацентарная продукция простациклинов и тромбоксанов в динамике физиологической и осложненной беременности // Росс. вестн. акуш-гинекол. 2008. Т. 8. № 3. С. 9-11.
8. Люсов В.А., Метельская В.А., Оганов Р.Г. и др. Суточная продукция N0 у больных артериальной гипертонией II стадии // Рос. кард. журн. 2001. № 6. С. 34-37.
9. Люсов В.А., Метельская В.А., Оганов Р.Г. и др. Уровень оксида азота в сыворотке периферической крови больных с различной тяжестью артериальной гипертензии // Кардиология. 2011. № 12. С. 23-28.
10. Максименко А.В., Турашев А.Д. Эндотелиальный гликокаликс системы кровообращения. I. Обнаружение, компоненты, структурная организация // Биоорганическая химия. 2014. Т. 40. № 2. С. 131-141.
11. Малинин В.В., Дурнова А.О., Полякова В.О. Факторы роста и молекулы адгезии эндотелия сосудов как молекулярные мишени для создания пептидных лекарственных препаратов против атеросклероза // Молекулярная медицина. 2013. №3. С. 53-55.
12. Марков Х.М. Оксид азота и атеросклероз. Оксид азота, дисфункция сосудистого эндотелия и патогенез атеросклероза // Кардиология. 2009. № 11. С. 64-74.
13. Петрищев Н. Н., Беркович О. А., Власов Т. Д. и др. Диагностическая ценность определения десквамированных эндотелиальных клеток в крови // Клин. лаборат. диагностика. 2001. № 1. С. 50-52.
14. Петрищев Н.Н., Васина Л.В., Власов Т.Д. и др. Типовые формы дисфункции эндотелия // Клинико-лабораторный консилиум. 2007. № 18. С. 31-35.
15. Петрищев Н.Н., Васина Л.В., Сапегин А.А. и др. Диагностическая значимость определения содержания факторов повреждения эндотелия для оценки выраженности эндотелиальной дисфункции при остром коронарном синдроме // Клиническая больница. 2015. Т. 1. № 11. С. 41-45.
16. Руда М.М., Арефьева Т.И., Соколова А.В. и др. Циркулирующие предшественники эндотелиальных клеток при нарушенном углеводном обмене у больных ишемической болезнью сердца // Кардиология. 2010. № 1. С. 13-20.
17. Рябченко А.Ю. Долгов А.М., Денисов Е.Н. и др. Роль оксида азота и эндотелина-1 в развитии ишемических нарушений мозгового кровообращения // Неврологический вестник. 2014. № 1. С. 34-37.
18. Скворцов Ю.И., Королькова А.С. Гомоцистеин как фактор риска развития ИБС (обзор) // Саратовский научно-медицинский журнал. 2011. Т. 7. №3. С. 619-624.
19. Соколов Е.И., Гришина Т.И., Штин С.Р. Влияние фактора Виллебранда и эндотелина-1 на формирование тромботического статуса при ишемической болезни сердца // Кардиология. 2013. №3. С. 25-30.
20. Соколов Е.И., Манухин И. Б., Мочалов А. А., Невзоров О. Б. Нарушение в системе гемостаза и его коррекция у беременных с метаболическим синдромом // Лечащий врач. 2011. № 3. С. 43-47.
21. Чернеховская Н.Е., Шишло В.К., Поваляев А.В., Шевхужев З.А. Коррекция микроциркуляции в клинической практике: монография. Москва: Бином, 2013. 208 с.
22. Шевченко А.О., Шевченко О.П., Эль-Бустани С., Князев А.Н. Маркер неоартериогенеза PLGF коррелирует с толщиной комплекса интима-медиа общей сонной артерии и отдаленным прогнозом у больных ИБС // Кардиоваскулярная терапия и профилактика. 2007. Т. 6. № 5. С. 340-341.
23. Aird W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms // Circ Res. 2007. Vol. 100 (2). P. 158-173. DOI: 10.1161/01. RES.0000255691.76142.4a
24. Aird W.C. Discovery of the cardiovascular system: from Galen to William Harvey // J Thromb Haemost. 2011; Vol. 9 (Suppl 1): P. 118-129. DOI: 10.1111/j.1538-7836.2011.04312.x
25. Alegret J.M, Martmez-Micaelo N., Aragones G., Beltran-Debon R. Circulating endothelial microparticles are elevated in bicuspid aortic valve disease and related to aortic dilation // Int. J. Cardiol. 2016. Vol. 15. № 217. Р. 35-41. DOI: 10.1016/j.ijcard.2016.04.184
26. Andrae J., Gallini R., Betsholtz C. Role of platelet-derived growth factors in physiology and medicine // Genes Dev. 2008. Vol. 22. № 10. Р. 1276-1312. DOI: 10.1101/gad.1653708
27. Anggrahini D.W., Emoto N., Nakayama K. et al. Vascular endothelial cell-derived endothelin-1 mediates vascular inflammation and neointima formation following blood flow cessation // Cardiovasc. Res. 2009. Vol. 82. P. 143-151. DOI: 10.1093/cvr/cvp026
28. Bach F.H., Hancock W.W., Ferran C. Protective genes expressed in endothelial cells: a regulatory response to injury // Immunol. Today. 1997. Vol. 18. P. 483-486. DOI: http://dx.doi.org/10.1016/S0167-5699(97)01129-8
29. Bach F.H., Robson S.C., Ferran C. et al. Endothelial cell activation and thromboregulation during xenograft rejection // Immunol. Rev. 1994. Vol. 141. P. 5-30. DOI: 10.1111/j.1600-065X.1994.tb00870.xView
30. Ballermann B.J. Endothelial cell activation // Kidney Int. 1998. Vol. 53. P. 1810-1826. DOI: 10.1046/j.1523- 1755.1998.00943.x
31. Bamashmoos S.A., Al-Nuzaily M.A., Al-Meeri A.M., Ali F.H. Relationship between total homocysteine, total cholesterol and creatinine levels in overt hypothyroid patients // Springerplus. 2013. Vol. 2. P. 423. doi: 10.1186/2193-1801- 2-423.
32. Blankenberg S., Rupprecht H.J., Bickel C. et al. Circulating cell adhesion molecules and death in patients with coronary artery disease // Circulation. 2001. Vol. 104. P. 1336-1341. https://doi.org/10.1161/hc3701.095949
33. Blann A.D. Endothelial cell activation, injury, damage and dysfunction: separate entities or mutual terms? // Blood Coagul. Fibrinolysis. 2000. Vol. 11. № 7. P. 623-630.
34. Blann A.D., Lip G.Y. Cell adhesion molecules in cardiovascular disease and its risk factors — what can soluble levels tell us? // J. Clin. Endocrinol. Metab. 2000. Vol. 85. № 5. P. 1745-1747. DOI: 10.1210/jcem.85.5.6594
35. Boffa G.M., Zaninotto M., Bacchiega E. et al. Correlations between clinical presentation, brain natriuretic peptide, big-endothelin, tumor necrosis factor-alpha and cardiac troponins in heart failure patients // Ital. Heart J. 2005. Vol. 6. № 2. P. 125-132.
36. Boneu B., Abbal M., Plante J., Bierme R. Factor VIII complex and endothelial damage // Lancet. 1975. Vol. 1. № 7922. P. 1430. DOI: http://dx.doi.org/10.1016/S0140-6736(75)92650-1
37. Bras-Silva C., Leite-Moreira A. F. Myocardial effects of endothelin-1 // Pev. Port. Cardiol. 2008. Vol. 27. P. 925-951.
38. Cipollone F., Marini M., Fazia M. et al. Elevated circulating levels of monocyte chemoattractant protein-1 in patients with restenosis after coronary angioplasty // Arterioscler. Thromb. Vasc. Biol. 2001. Vol. 21. P. 327-34. https://doi.org/10.1161/01.ATV2L3.327
39. Collet J., Montalescot G., Vicaut E. et al. Acute release of plasminogen activator inhibitor-1 in ST-segment elevation myocardial infarction predicts mortality // Circulation. 2003. Vol. 108. P. 391-94. https://doi.org/10.1161/01. CIR.0000083471.33820.3C
40. Collins S.R., Blank R.S., Deatherage L.S. The endothelial glycocalyx: emerging concepts in pulmonary edema and acute lung injury // Anesth. Analg. 2013. Vol. 117. P. 664-674. DOI: 10.1213/ANE.0b013e3182975b85
41. Cotran R.S. New roles for the endothelium in inflammation and immunity // Am. J. Pathol. 1987. Vol. 129. P. 407-13.
42. Cotran R.S., Gimbrone M.A., Bevilacqua M.P., et al. Induction and detection of a human endothelial activation antigen in vivo // J. Exp. Med. 1986. Vol. 164. P. 661-666. DOI: 10.1084/jem.164.2.661
43. Davis K.L., Martin E., Turko I.V., Murad F. Novel effects of nitric oxide // Annu. Rev. Pharmacol. Toxicol. 2001. Vol. 41. P. 203-236. DOI: 10.1146/annurev.pharmtox.41.1.203
44. De La Cruz J.P., Villalobos M.A., Escalante R. et al. Effects of the selective inhibition ofplatelet thromboxane synthesis on the platelet-subendothelium interaction // Br. J. Pharmacol. 2002. Vol. 137. P. 1082-88. doi: 10.1038/sj.bjp.0704963
45. Dignat-George F., Boulanger C.M. The many faces of endothelial microparticles // Arterioscler. Thromb. Vasc. Biol. 2011. Vol. 31. P 27-33. DOI: 10.1161/ATVBAHA.110.218123
46. Dignat-George F., Sampol J., Lip G.Y., Blann A.D. Circulating endothelial cells: realities and promises in vascular disorders // Pathophysiol. Haemost. Thromb. 2004. Vol. 33. Р. 495-499. DOI: 83851
47. Durand M.J., Gutterman D.D. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease // Microcirculation. 2013. Vol. 20. № 3. P 239-247. DOI: 10.1111/micc.12040
48. Ebong E.E., Macaluso F.P., Spray D.C., Tarbell J.M. Imaging the endotelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy // Arterioscler. Thromb. Vasc. Biol. 2011. Vol. 31. № 8. P. 1908-1915. DOI: 10.1161/ATVBAHA.n1.225268
49. Endemann D.H., Schiffrin E.L. Endothelial dysfunction // J. Am. Soc. Nephrol. 2004. Vol. 15. № 8. Р. 1983-1992. DOI: 10.1097/01.ASN.0000132474.50966.DA
50. Florey H.W. The endothelial cell // Br. Med. J. 1966. Vol. 2. № 5512. Р. 487-490.
51. Garlanda C., Dejana E. Heterogeneity of endothelial cells. Specific markers // Arterioscler. Thromb. Vasc. Biol. 1997. Vol. 17. № 7. P. 1193-202. https://doi.org/10.1161/01. ATV 17.7.1193
52. George E.M., Granger J.P. Endothelin: key mediator of hypertension in preeclampsia // Am. J. Hypertens. 2011. Vol. 24. № 29. P. 964-969. DOI: 10.1038/ajh.2011.99
53. Goepfert C., Imai M., Brouard S. et al. CD39 modulates endothelial cell activation and apoptosis // Mol. Med. 2000. Vol. 6. № 7. P. 591-603.
54. Goon P.K., Boos C.J., Lip G. Y. Circulating endothelial cells: markers of vascular dysfunction // Clin. Lab. 2005. Vol. 51. Р. 531-538.
55. Goon P.K., Lip G.Y., Boos C.J. Circulating endothelial cells, endothelial progenitor cells, and endothelial microparticles in cancer // Neoplasia. 2006. Vol. 8. Р. 79-88. DOI: 10.1593/neo.05592
56. Granger D.L., Anstey N.M., Miller W.C., Weinberg J.B. Measuring nitric oxide production in human clinical studies // Methods Enzymol. 1999. Vol. 301. P. 49-61. http:// dx.doi.org/10.1016/S0076-6879(99)01068-X
57. Hwang S.-J., Ballantyne S.M., Sharrett R. et al. Circulating adhesion molecules VCAM-1, ICAM-1 and E-selectin in carotid atherosclerosis and incident coronary heart disease cases. The Atherosclerosis Risk in Communities (ARIC) study // Circulation. 1997. Vol. 96. P.4219-4225. https:// doi.org/10.1161/01.CIR.96.12.4219
58. Ikeda H., Nakayama H., Oda T. et al. Neutrophil activation after percutaneous transluminal coronary angioplasty // Am. Heart J. 1994. Vol.128. P. 1091-1098.
59. Jansson, J., Nilsson T., Johnson O. Von Willebrand factor in plasma: a novel risk factor for recurrent myocardial infarction and death // Br. Heart J. 1991. Vol. 66. P. 351-355.
60. Kalinina N., Agrotis A., Antropova Y. Smadexpression in human atherosclerotic lesions: evidence for impaired TGF-beta/Smad signaling in smooth muscle cells of fibrofatty lesions // Arterioscler. Thromb. Vasc. Biol. 2004. Vol. 24. № 8. Р. 1391-1396. DOI: 10.1161/01.ATV0000133605.89421.79
61. Katrinchak C., Fritz K. Clinical implications of C-reactive protein as a predictor of vascular risk // J. Am. Acad. Nurse Pract. 2007. Vol. 19. № 7. Р. 335-340. DOI: 10.1111/j.1745-7599.2007.00234.x
62. Kawamoto A., Asahara T. Role of progenitor endothelial cells in cardiovascular disease and upcoming therapies // Catheter Cardiovasc. Interv. 2007. Vol. 70(4). P. 477-84. DOI: 10.1002/ccd.21292
63. Khan S.S., Solomon M.A., McCoy J.P. Detection of circulating endothelial cells and endothelial progenitor cells by flow cytometry // J. Cytometry. B. Clin. Cytom. 2005. Vol. 64. № 1. Р. 1-8. DOI: 10.1002/cyto.b.20040
64. Kitchens C.S., Konkle B.A., Kessler C.M. Consultative hemostasis and thrombosis, 3rd ed. Philadelphia.: Elsevier/Saunders, 2013. 840 p.
65. Koo A., Dewey C.F. Jr., Garcia-Cardena G. Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyxformation in cultured endothelial cells // Am. J. Physiol. Cell Physiol. 2013. Vol. 304. № 2. Р. 137-146. doi: 10.1152/ajpcell.00187.2012
66. Lip G.Y.H., Lowe G.D.O., Metcalfe M.J. et al. Effects of warfarin therapy on plasma fibrinogen, von Willebrand factor and fibrin D-dimer in left ventricular dysfunction secondary to coronary artery disease with and without aneurysms // Amer. J. Cardiol. 1995. Vol. 76. P. 453-8.
67. Lip G.Y.H., Lowe G.D.O., Rumley A., Dunn F.G. Increased markers of thrombogenesis in chronic atrial fibrillation: effects of warfarin therapy // Br. Heart J. 1995. Vol. 73. № 6. P. 527-533.
68. Malik I.S., Hascard D.O. Soluble adhesion molecules in ishaemic heart disease // Eur. Heart J. 1999. Vol. 20. № 14. P.990-991. DOI: 10.1053/euhj.1999.1576
69. Martmez M.C., Tesse A., Zobairi F., Andriantsitohaina R. Shed membrane microparticles from circulating and vascular cells in regulating vascularfunction // Am. J. Physiol. Heart Circ. Physiol. 2005. Vol. 288. № 3. Р. 1004-1009. DOI: 10.1152/ajpheart.00842.2004
70. Masaoka H. Suzuki R., Hirata Y. et al. Raised plasma endothelin in aneurysmal subarachnoid haemorrhage // Lancet. 1989. Vol. 2. P. 1402. DOI: http://dx.doi.org/10.1016/ S0140-6736(89)92019-9
71. Miranda K.M., Espey M.G., Wink D. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite // Nitric Oxide. 2001. Vol. 5. № 1. P. 62-71. DOI: 10.1006/niox.2000.0319
72. Montoro-Garda S., Shantsila E., Lip G.Y.H. Potential value of targeting von Willebrand factor in atherosclerotic cardiovascular disease // Expert Opin. Ther. Targets. 2014. Vol. 18. № 1. Р. 43-53. DOI: 10.1517/14728222.2013.840585
73. Morisaki N., Saito I., Tamura K. et al. New indices of ischemic heart disease and aging: studies on the serum levels of soluble intercellular adhesion molecule-1 and soluble vascular cell adhesion molecule-1 in patients with hypercholesterinemia and ischemic heart disease // Atherosclerosis. 1997. Vol. 131. № 1. P. 43-48. DOI: http://dx.doi. org/10.1016/S0021-9150(97)06083-8
74. Neves F.M., Meneses G.C., Sousa N.E. et al. Syndecan-1 in acute decompensated heart failure-association with renal function and mortality // Circ. J. 2015. Vol. 79. № 7. Р. 1511-19. http://doi.org/10.1253/circj.CJ-14-1195
75. Olson N.E., Chao S., Lindner V., Reidy M.A. Intimal smooth muscle cell proliferation after balloon catheter injury: the role of basicfibroblast growth factor // Am. J. Pathol. 1992. Vol. 140. P. 1017-1023.
76. Padberg J.S., Wiesinger A., di Marco G.S. et al. Damage of the endothelial glycocalyx in chronic kidney disease // Atherosclerosis. 2014. Vol. 234. № 2. Р. 335-43. doi: 10.1016/j.atherosclerosis.2014.03.016.
77. Pizarro S., Garda-Lucio J., Peinado V.I. et al. Circulating progenitor cells and vascular dysfunction in chronic obstructive pulmonary disease // PLoS One. 2014. Vol. 9. № 8. e106163. doi: 10.1371/journal.pone.0106163.
78. Pober J.S. Activation and injury of endothelial cells by cytokines // Pathol. Biol. 1998. Vol. 46. P. 159-163.
79. Pober J.S., Cotran R.S. Cytokines and endothelial cell biology // Physiol. Rev. 1990b. Vol. 70. P. 427-451.
80. Przybylski M. A review of the current research on the role of bFGF and VEGF in angiogenesis // J. Wound Care. 2009. Vol. 18. № 12. Р. 516-519. DOI: 10.12968/ jowc.2009.18.12.45609
81. Quilici J., Banzet N., Paule P. et al. Circulating endothelial cell count as a diagnostic marker for non-ST-elevation acute coronary syndromes // Circulation. 2004. Vol. 110. № 12. P. 1586-1591. DOI: 10.1161/01. CIR.0000142295.85740.98
82. Rajendran P., Rengarajan T., Thangavel J. et al. The vascular endothelium and human diseases // Int. J. Biol. Sci. 2013. Vol. 9. № 10. Р. 1057-69. DOI: 10.7150/ijbs.7502
83. Stumpf С., Jukic J., Yilmaz A. et al. Elevated VEGF-plasma levels in young patients with mild essential hypertension // Eur. J. Clin. Invest. 2009. Vol.39. № 1. Р. 31-6. DOI: 10.1111/j.1365-2362.2008.02056.x
84. van den Berg B.M., Spaan J.A., Rolf T.M., Vink H. Atherogenetic region and diet diminish glycocalyx dimension and increase intima-to-media rations at murine carotid artery bifurcation // Am. J. Physiol. Heart Circ. Physiol. 2006. Vol. 290. № 2. H915-20. DOI: 10.1152/ajpheart.00051.2005
85. Vita J.A., Keaney J.F. Endothelial function // Circulation. 2011. Vol. 124. № 25. Р. e906-12. https://doi.org/10.1161/01.CIR.0000028581.07992.56
86. Vlahu C.A., Lemkes B.A., Struijk D.G., et al. Damage of the endothelial glycocalyx in dialysis patients // J. Am. Soc. Nephrol. 2012. Vol. 23. № 11. Р. 1900-1908. DOI: 10.1681/ASN.2011121181
87. Weinbaum S., Tarbell J.M., Damiano E.R. The structure and function of the endothelial glycocalyx layer // Annu. Rev. Biomed. Eng. 2007. Vol. 9. P. 121-167. DOI: 10.1146/ annurev bioeng.9.060906.151959
88. Werner N., Wassmann S., Ahlers P. et al. Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease // Arterioscler. Thromb. Vasc. Biol. 2006. Vol. 26. № 1. Р. 112-116. DOI: https://doi.org/10.1093/eurheartj/ehq478
89. Wie C.M., Lerman A., Rodeheffer R.J. et al. Endothelin in human congestive heart failure // Circulation. 1994. Vol. 89. P. 1580-1586. https://doi.org/10.1161/01. CIR.89.4.1580
90. Willey K.E., Davenport A.P. Nitric oxide-medulation of the endothelin-1 signaling pathway in the human cardiovascular system // Br. J. Pharmacol. 2001. Vol. 132. P. 213-220. doi: 10.1038/sj.bjp.0703834
91. Woolf N. The arterial endothelium. In: Crawford ST, ed. Pathology of Atherosclerosis. London, England: Butterworths & Co Ltd, 1982. Р. 25-45.
92. Wu H., Chen H., Hu P.C. Circulating endothelial cells and endothelial progenitors as surrogate biomarkers in vascular dysfunction // Clin. Lab. 2007. Vol. 53. № 5-6. Р. 285-295.
93. Yetgin S., Yenicesu I., Cetin M., Tuncer M. Clinical importance of serum vascular endothelial and basicfibroblast growth factors in children with acute lymphoblastic leukemia // Leuk. Lymphoma. 2001. Vol. 42. № 1-2. Р. 83-88. DOI: 10.3109/10428190109097679
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Дзугкоев С.Г.
1
Можаева И.В.
1
Такоева Е.А.
1
Дзугкоева Ф.С.
1
Маргиева О.И.
1
1 Институт биомедицинских исследований Владикавказского научного центра РАН и Правительства РСО-Алания
В обзоре приведены данные литературы, свидетельствующие о механизмах развития эндотелиальной дисфункции при сосудистых осложнениях сахарного диабета, артериальной гипертензии, ИБС, атеросклерозе. Установлено, что ключевую роль играет нарушение метаболизма оксида азота, понижение содержания суммарных метаболитов оксида азота вследствие снижения уровня экспрессии eNOS, а также нарушения биодоступности оксида азота. В механизме сниженной эффективности оксида азота играет роль гиперхолестеринемия, гипер-β-липопротеинемия и атерогенные изменения в сосудистой стенке. С другой стороны, вносит свой вклад аргиназа – фермент цикла синтеза мочевины. Анализ литературных данных и собственных исследований открывает возможность коррекции нарушений метаболизма оксида азота путем ингибирования фермента аргиназы и введением донора оксида азота – L-аргинина и их комбинации.
эндотелиальная дисфункция
eNOS
оксид азота
аргиназа
L-аргинин
1. Бабак О.Я. Артериальная гипертензия и ишемическая болезнь сердца – эндотелиальная дисфункция: современное состояние вопроса / О.Я. Бабак, Ю.Н. Шапошников, В.Д. Немцова // Укр. Терапевт.журн. – 2003. – № 1. – С. 14–21.
2. Голиков П.П. Динамика оксида азота (NOx) и других факторов окислительного стресса в прецеребральных сосудах до и после каротидной эндартерэктомии / П.П. Голиков, В.Л. Леменев, В.В. Ахметов и др. // Регионар. кровообращ. и микроцирк. – 2003. – № 4. – С. 28–33.
3. Головченко Ю.И. Обзор современных представлений об эндотелиальной дисфункции / Ю.И. Головченко, М.А. Трещинская // Consilium medicum Ukraina. – 2008. – № 11. – С. 38–40.
4. Дзугкоев С.Г. Влияние конзима Q10, афобазола и L- карнитина и их комбинации с L- аргинином на эндотелиальную функцию у крыс с ЭСД / С.Г. Дзугкоев, Ф.С. Дзугкоева, Н.Г. Гуманова, В.А. Метельская // Вопросы биологической медицинской и фармацевтической химии. – 2012. – № 11. – С. 49–53.
5. Дзугкоев С.Г. Влияние L-аргинина на показатели функции эндотелия при сахарном диабете в эксперименте / С.Г. Дзугкоев, Д.М. Никулина, И.В. Можаева // 9-ая международная научно-практическая конференция «Достижения фундаментальных наук и возможности трансляционной медицины в решении актуальных проблем практического здравоохранения». (Астрахань, 6–8 мая 2013 г). – 2013. – С. 83–84.
6. Корокин М.В. Влияние L-аргинина, витамина В6 и фолиевой кислоты на показатели эндотелиальной дисфункции и микроциркуляции в плаценте при моделировании L-NAME-индуцированного дефицита оксида азота / М.В. Корокин, М.В. Покровский, О.О. Новиков, В.В. Гуреев, Т.А. Денисюк, Л.В. Корокина, О.С. Полянская, В.А. Рагулина, Т.Г. Покровская, Л.М. Даниленко, А.С. Белоус // Бюллетень экспериментальной биологии и медицины. – 2011. – Т. 152. – № 7. – С. 77–79.
7. Коркушко О.В. Эндотелиальная дисфункция / О.В. Коркушко, В.Ю. Лишневская // Кровообiг та гемостаз. – 2003. – № 2. – С. 4–15.
8. Ланкин В.З. Антиоксиданты в комплексной терапии атеросклероза: pro et contra / В.З. Ланкин, А.К. Тихадзе, Ю.Н. Беленков // Кардиология. – 2004. – Т.44. – № 2. – С. 72–81.
9. Марков Х.М. L-аргинин – оксид азота в терапии болезней сердца, и сосудов // Кардиология. – 2005. – Т. 45, № 6. – С. 87–95.
10. Марков Х.М. Оксид азота и сердечно-сосудистая система // Успехи физиолог, наук. – 2001. – Т. 32, № 3. – С. 9–65.
11. Марков Х.М. Оксидантный стресс и дисфункция эндотелия // Патол. физиология и эксперим. терапия. – 2005. – № 4. – С. 59.
12. Небиеридзе Д.В. Клиническое значение дисфункции эндотелия при артериальной гипертонии // Consilium medicum: Системные гипертен. (прил.). – 2005. – Т. 7, № 1. – С. 31–38.
13. Тюренков И.Н. Методический подход к оценке эндотелиальной дисфункции в эксперименте / И.Н. Тюренков, А.В. Воронков // Эксперим. и клин. фармакология. – 2008. – Т. 71, № 1. – С. 49–51.
14. Шестакова М.В. Дисфункция эндотелия причина или следствие метаболического синдрома // Рус. мед. журн. – 2001. – Т. 9, № 2. – С. 88–90.
15. Barbato J.E. Tzeng E. Nitric oxide and arterial disease // J. Vase. Surg – 2004. – Vol. 40. – P. 187–193.
16. Berkowitz D.E., White R., Li D., et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. – 2003. – 108. – Р. 2000–6.
17. Boger R.N., Bode-Boger S.M., Szuba A. et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. – 1998. – Vol. 98. – P. 1842–1847.
18. Boger R.H., Bode-Boger S.M. The clinical pharmacology of L-arginine // Annu Rev Pharmacol Toxicol. – 2001. – Vol.41. – P. 79–99.
19. Brodiak I.V., Sybirna N.O. Peculiarities of L-arginine metabolism in the blood leukocytes in experimental diabetes mellitus // Fiziol Zh. – 2008. – № 54(1). – P. 63–68.
20. Chiu J.J., Usami S., Chien S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis // Ann. Med. – 2008. – Vol. 18. – P. 1–10.
21. Cooke J.P. Losordo D.W. Nitric oxide and angiogenesis. Circulation. 2002. – № .105. – P. 2133–2138.
22. Costanzo D.L., Ilies M., Thorn K.J., Christianson D.W. Inhibition of human arginase I by substrate and product analogues // Arch. Biochem. Biophys. – 2010. – № 496 (2). – P. 101–108.
23. Costanzo L.D., Sabio G., Mora A. et al. Crystal structure of human arginase I at 1.29-A resolution and exploration of inhibition in the immune response // PNAS. – 2005. – Vol.102. – № 37. – P. 13058–13063.
24. Durante W., Johnson F.K., Johnson R.A. Arginase: a critical regulator of nitric oxide synthesis and vascular func tion // Clinical and Experimental Pharmacology and Physio logy. – 2007. – Vol. 34(9). – P. 906–911.
25. Fike C.D., Kaplowitz M.R., Rehorst-Paea L.A. et al. L-arginine increases nitric oxide production in isolated lungs of chronically hypoxic newborn pigs // J. Appl. Physiol. – 2000. – Vol. 88. – № 5. – P.1797–1803.
26. Forstermann U., Boissle J.-P., Kleinert Н. Expressional control of the «constitutive» isoforms of nitric oxide synthase (NOS I and NOS III) // FASEB J. – 1998. – № 12. – P. 773–90.
27. Ghisi G.L., Durieux A., Pinho R., Benetti H. Physical exercise and endothelial dysfunction // Arg. Bras. Cardiol. – 2010. – Vol. 95. – № 5. – P. 130–137.
28. Giraldelo C.M., Zappellini A., Muscara M.N. et al. Effect of L-arginine anologues on rat hind paw oedema and mast cell activation in vitro // Eur J Pharmacol. – 1994. – № 257. – P. 87–93.
29. Gronros J., Jung C., Lundberg J.O. Arginase inhibition restores in vivo coronary microvascular function in type 2 diabetic rats. // Am J Physiol. Heart Circ. Physiol. – 2011. – Vol. 300. – № 4. – P. l174–1181.
30. Guiliano D., Marfella R., Verazzo G. et al. The vascular effects of L-arginine in humans: the role of endogenous insulin // J Clin Invest. – 1997. – № 99. – P. 433–438.
31. Herman A.G., Moncada S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis // Eur. Heart. J. – 2005. – Vol. 26(19). – P. 1945–1955.
32. Huynh N.N., Harris E.E., Chin-Dusting J.F., And rews K.L. The vascular effects of different arginase inhibitors in rat isolated aorta and mesenteric arteries // British Journal of Pharmacology. – 2009. – № 156. – P. 84–93.
33. Jay M.T., Chirico S., Siow R.C., et al. Modulation of vascular tone by low density lipoproteins: effects on L-arginine transport and nitric oxide synthesis // Exp Physiol. – 1997. – № 82. – P. 349–60.
34. Jeyabalan G., Klune J.R., Nakao A. et al. Arginase blockade protects against hepatic damage in warm ischemia-reperfusion, Nitric Oxide. – 2008. – Vol.19. – P. 29–35.
35. Johnson F.K., Johnson R.A., Peyton K.J., Durante W.. Arginase inhibition restores arteriolar endothelial function in Dahl rats with salt-induced hypertension // Am J Physiol. Regul. Integr. Comp. Physiology. – 2005. – Vol. 288. – P. 1057–1062.
36. Jung C., Gonon A.T., Sjoquist P.O. et al. Arginase inhibition mediates cardioprotection during ischaemia-reperfusion // Cardiovascular Research. – 2010. – Vol.85(1). – P. 147–154.
37. Kielstein J.T., Impraim B., Simmel S., et al. Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans // J. C. Circulation. – 2004 – Vol. 109. – P. 172–177.
38. Kim J.H., Bugaj L.J, Oh Y.J. et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats // J Appl. Physiol. – 2009. – Vol. 107(4). – P. 1249–1257.
39. Luiking Y.C., Engelen M.P., Deutz N.E. Regulation of nitric oxide production in health and disease // Curr. Opin. Clin. Nutr. Metab. Care. – 2010. – Vol. l3(l). – P. 97–104.
40. Maarsingh H., Zuidhof A.B., Bos I.S. et al. Arginase inhibition protects against allergen-induced airway obstruction, hyperresponsiveness, and inflammation // Am J Respir. Crit. Care Med. – 2008. – Vol. 178(6). – P. 565–573.
41. Michael T., Gewaltig M., Kojda G. Vasoprotection by nitric oxide: mechanisms and therapeutic potential // Cardiovascular research. – 2002. – Vol. 55. – P. 205–260.
42. Miller A.L. The effects of sustained-release-L-arginine formulation on blood pressure and vascular compliance in 29 healthy individuals // Altern. Med. Rev. – 2006. – Vol. 11(1). – P. 23–29.
43. Morris C.R., Morris S.M., Hagaret Jr., W. et al. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease // Am. J. Respir. Crit. Care. Med. – 2003. – Vol. 168. – № 1. – P. 63–69.
44. Nigris F., Lerman L.O., Ignarro S.W. et al. Beneficial effect of antioxidants and L-arginine on oxidant-sensitive gene expression and endothelial NO synthase activity at sites of disturbed shear stress // PNAS. – 2003. – Vol. 100. – P. 1420–1425.
45. Rabelink T.J., Luscher T.F. Endothelial nitric oxide synthase host defense enzyme of the endothelium? Arterioscl // Throm., and Vascular Biology. – 2006. – Vol. 26. – P. 267–271.
46. Santhanam L., Christianson D.W., Nyhan D., Berko witz D. E Arginase and vascular aging // J Appl. Physiology. – 2008. – Vol. 105. – P. 1632–1642.
47. Simon, A., Castro A., Kaski J.C. Assessment of endothelial dysfunction and its clinical usefulness, A. Simon // Rev. Esp. Cardiol. – 2001. – Vol. 54. – P. 2117.
48. Taddei S, Virdis А, Matter Р, et аl. Defective L-arginine-nitric oxide pathway in offspring of essentia1 hypertensive patients // Circulation. – 1996. – Vol. 94. – P. 1296–1303.
49. Taddei S., Virdis A., Ghiadoni L. et. al. Endothelium, aging, and hypertension // Curr. Hypertens. Rep. – 2006. – Vol. 8. – № 1. – P. 84–89.
Известно, что заболевания сердечно-сосудистой системы ежегодно уносят жизни миллионов человек по всему миру. В настоящее время накоплено достаточно большое количество исследований, демонстрирующих связь между нарушением функции эндотелия и сердечно-сосудистой патологией [1, 7, 22, 35]. В патогенезе и клинике сосудистых осложнений сахарного диабета, артериальной гипертензии, ишемической болезни сердца, атеросклерозе играет роль нарушение структурно-функциональной организации сосудистого эндотелия [10, 14, 11, 9, 35, 49]. Многочисленными исследованиями установлено, что эндотелий регулирует сосудистую проницаемость, выделяя вазоактивные вещества [13, 36], т.е. эндотелиоциты выполняют важнейшую метаболическую функцию, участвуют в регуляции сосудистого тонуса, адгезии лейкоцитов, тромбоцитов, роста сосудов, иммунобиологических свойств. Среди медиаторов, синтезируемых эндотелием, выделяют вазоконстрикторы (эндотелин І, ангиотензин II) и вазодилататоры (эндотелиальный гиперполяризующий фактор, простациклин и оксид азота (NО)) [37, 19, 36]. Дисбаланс между вазодилататорами и вазоконстрикторами с преобладанием последних приводит к так называемой эндотелиальной дисфункции [47]. Основное внимание исследователей в последние годы привлекает молекула оксида азота, являющаяся вазодилатирующим фактором. В нормально функционирующем эндотелии низкие уровни N0 постоянно высвобождаются [12, 37]. Он образуется из условно незаменимой аминокислоты L-аргинина при участии NO-синтаз (NOS). Существуют три изоформы фермента NOS, две из которых постоянно функционирующие или конститутивные ферменты: нейрональная (NOS 1 или nNOS) и эндотелиальная (NOS 3 или еNOS) NO-синтазы [26]. Эти изоформы постоянно экспрессируются в нейронах и эндотелиальных клетках (ЭК), для их активации необходимы ионы Ca2+ и кальмодулин [7]. Индуцибельная NOS (NOS 2 или iNOS) – это фермент, который экспрессируется и функционирует в ответ на действие эндотоксинов и провоспалительных цитокинов, бактериальных липополисахаридов, для ее активации практически не требуются ионы Ca2+ [7]. NO-синтазы осуществляют присоединение молекулярного кислорода к атому азота из терминальной гуанидиновой группы L-аргинина. В этой ферментной реакции участвует ряд вспомогательных факторов, включая никотиновые и флавиновые коферменты, тетрагидробиоптерин, ионы кальция, кальмодулин. NO образуется в организме при восстановлении неорганических нитратов и нитритов. Проникая из эндотелиальных клеток в гладкомышечные клетки (ГМК) сосудистой стенки, он активирует гем-содержащий фермент – растворимую гуанилатциклазу, в результате в ГМК повышается уровень циклического гуанозинмонофосфата (цГМФ) и, соответственно, цГМФ-зависимых протеинкиназ, снижается концентрация Ca2+ и происходит расслабление сосудов [5]. Период полураспада для оксида азота исчисляется несколькими секундами в условиях in vitro и сотыми долями секунды in vivo.
Ключевым звеном формирования эндотелиальной дисфункции (ЭД) считают снижение уровня биологически активного NO, продуцируемого сосудистым эндотелием, или снижение его биодоступности. Важнейшим звеном в цепи механизмов, ведущих к развитию дефицита NO и ЭД при многих патологиях, в том числе и при сахарном диабете (СД), является дефицит концентрации L-аргинина, которая колеблется в зависимости от возраста и диеты. В норме концентрация этой аминокислоты составляет 45–150 мкмоль/л в сыворотке крови [18]. Количество проникающего в эндотелиальные клетки аргинина зависит от активности мембранно-связанной транспортной системы, активности аргиназы и других факторов. Повышение концентрации L-аргинина в эндотелиальных клетках сопровождается усиленным образованием промежуточного продукта метаболизма аргинина – NG-гидрокси-L-аргинина, облегчающего процесс окисления аргинина NO-синтазой, а также тормозящего активность аргиназы [6,16,19]. С другой стороны, аргинин увеличивает секрецию инсулина [28, 30], повышает синтез NO, активность eNOS, вызывая эндотелийзависимую вазодилатацию. Поступление L-аргинина в ЭК подвержено негативному влиянию окисленных липопротеинов низкой плотности (оЛНП) и лизофосфатидилхолина, продукта реакции, катализируемой фосфолипазой, ассоциированной с липопротеинами низкой плотности (ЛНП) [11, 21, 33]. ЛНП тормозят высвобождение NО эндотелиальными клетками и уменьшают эндотелийзависимую вазодилатацию [2, 1].
Второй причиной низкой биодоступности NO может быть повышенный уровень в плазме крови ассиметричного диметиларгинина (АДМА), эндогенного конкурентного ингибитора еNOS, который препятствует нормальной продукции NO и может стать причиной развития дисфункции эндотелия и сосудистой инсулинорезистентности. Еще один механизм снижения продукции NO связан с повышенной продукцией активных форм кислорода (АФК) и способностью NO взаимодействовать с ними. Это касается и сниженного уровня продукции NO при СД. [4]. В некоторых случаях может иметь место и генетическая природа снижения экспрессии еNOS, о чем свидетельствуют структурные изменения (полиморфизм) еNOS-гена у больных эссенциальной артериальной гипертензией, а также снижение уровня экспрессии NO-синтазы и эндотелийзависимой вазодилатации у подростков с первичной артериальной гипертензией задолго до ее клинических проявлений [48].
Известно, что метаболизм L-аргинина в клетках протекает двумя путями [29]. L-аргинин посредством аргиназы гидролизуется в орнитин и мочевину. Другой путь превращения L-аргинина в оксид азота и цитруллин катализируется NO-синтазой. Ферменты аргиназа и NO-синтаза конкурируют между собой за общий субстрат – L-аргинин [44, 15, 17, 40, 46].
Аргиназа, фермент цикла синтеза мочевины, обладает высокой активностью, превышает таковую NO-синтазы [15]. Она представлена в виде двух изоформ: аргиназа I – печеночная форма и аргиназа II – внепеченочная форма [43, 32], локализующаяся чаще в почках, простате, тонкой кишке. Ряд исследователей подтверждает связь повышенной активности аргиназы с развитием ЭД [42, 45, 32, 39]. Причина этого заключается в том, что аргиназа ингибирует еNOS, препятствуя, таким образом, продукции оксида азота. Снижение активности аргиназы приводит к повышению выработки оксида азота, благоприятно воздействует на нормализацию сосудистой функции. Таким образом, использование ингибиторов аргиназы для увеличения синтеза NO является одним из методических подходов предотвращения развития дисфункции эндотелия. Ингибиторы аргиназы – это вещества природного происхождения, механизм действия которых заключается в блокировании фермента аргиназы, а, следовательно, в нарушении превращения L-аргинина в орнитин и мочевину [41, 42]. Вследствие этого большее количество L-аргинина расщепляется под действием NO-синтазы с образованием оксида азота. Ингибиторы аргиназы могут быть селективными и неселективными. Представителем их является L-норвалин – наименее изученный неселективный ингибитор аргиназы. По данным ряда современных авторов, повышение активности фермента цикла синтеза мочевины – аргиназы – наблюдается при многих патологиях, таких как СД, бронхиальная астма, гломерулонефриты, псориаз [34]. Рядом исследований установлено, что ингибирование этого энзима способствует увеличению продукции оксида азота и предотвращению дисфункциональных нарушений в эндотелии [35].
В единичных исследованиях продемонстрировано эндотелио- и кардиопротективное действие комбинации ингибитора аргиназы L-норвалина и донатора оксида азота L-аргинина, вводимого крысам внутрижелудочно, на фоне модели ЭД. В исследованиях ряда авторов были выбраны две модели патологии для оценки влияния на эндотелий изучаемых веществ: гипергомоцистеин-индуцированная дисфункция эндотелия и L-NАМЕ-индуцированная ЭД. L-NАМЕ – аналог эндогенного ингибитора АДМА в организме подавляет выработку N0 путем угнетения экспрессии фермента NOS-3. Дефицит оксида азота вызывает снижение эффектов эндотелийзависимых вазодилататоров и повышение вазоконстрикторных влияний, изменение системной и региональной гемодинамики, повышение артериального давления, нарушение функционального состояния миокарда, увеличение экспрессии адгезивных молекул эндотелия и другие изменения [3, 15, 38, 27]. В результате экспериментов с блокадой NО-синтазы с помощью L-NAME происходило развитие артериальной гипертензии. Кроме того, развивающийся дефицит оксида азота проявлялся увеличением коэффициента эндотелиальной дисфункции, снижением концентрации стабильных метаболитов оксида азота в сыворотке крови, гипертрофией миокарда, мышечного слоя сосудистой стенки. Однако при сравнительном анализе коэффициент эндотелиальной дисфункции у животных с гипергомоцистеин-индуцированной дисфункцией эндотелия был ниже, чем у животных с L-NAМЕ-индуцированной патологией. Эти данные заставляют задуматься о более выраженном нарушении соотношения эндотелийзависимой вазодилатации и эндотелийнезависимой вазодилатации при введении L-NAME, нежели при введении метионина.
Механизм действия L-норвалина объясняется его структурным подобием с орнитином, который является одним из продуктов метаболизма цикла синтеза мочевины. Опосредованное действие L-норвалина на активность аргиназы связано с ингибированием орнитинтранскарбамоилазы, которая катализирует превращение орнитина в цитруллин в цикле синтеза мочевины [20]. В результате происходит избыточное накопление орнитина, который, в свою очередь, ингибирует аргиназу. Более того, L-норвалин повышает эндогенный синтез L-аргинина из цитруллина за счет ингибирования аргининосукцинатсинтазы, что приводит также к возрастанию продукции N0. Эндотелиопротективные эффекты L-норвалина обусловлены подавлением активности аргиназы, за счет чего происходит увеличение продукции главного вазодилатирующего медиатора эндотелия – оксида азота. Удалось выявить предотвращение повышенной адренореактивности и снижение миокардиального резерва на фоне повышения прироста сократимости миокарда при проведении гипоксической пробы. Кардиопротекция L-норвалином, по нашему мнению, может быть связана с повышением эндогенного синтеза L-аргинина за счет ингибирования активности аргининосукцинатсинтазы [25], а также накоплением L-аргинина в связи с ингибированием аргиназы. В свою очередь L-аргинин обладает рядом свойств, благотворно влияющих на деятельность сердечно-сосудистой системы: способствует деполяризации мембран клеток эндотелия и регулирует рН в этих клетках, а также рН крови, способствует нормализации реологических свойств крови, снижает формирование свободных радикалов и обеспечивает удаление их из клеток эндотелия [23, 24]. Морфологические исследования демонстрируют предотвращение под влиянием L-норвалина увеличения поперечного диаметра миокардиоцитов у животных с моделированием дисфункции эндотелия введением L-NAME. А при изучении биохимических параметров сыворотки крови определили увеличение концентрации стабильных метаболитов оксида азота при L-NAМЕ- и гомоцистеин-индуцированной эндотелиальной дисфункции введением L-норвалина.
Комбинация L-норвалина и L-аргинина обладала кардиопротективным действием в обеих моделях дисфункции эндотелия. Сочетанное применение указанных веществ в более полном объеме предотвращало исчерпание миокардиального резерва при пробе на адренореактивность и нагрузку сопротивлением, а также развитие гипертрофии кардиомиоцитов. При монотерапии L-аргинином эндотелио- и кардиопротективные свойства значительно уступают сочетанному использованию L-аргинина и L-норвалина. Более ярко выраженные эффекты сочетанного применения L-аргинина и L-норвалина связаны с кумулятивными возможностями данной комбинации, с увеличением концентрации субстрата синтеза NO и ингибированием фермента аргиназы, L-норвалин увеличивает доступность эндогенных запасов L-аргинина для NО-синтазы, а также повышает активность фермента NOS-3. Введение экзогенного L-аргинина повышает концентрацию субстрата для eNOS. В исследованиях в нашей лаборатории установлено, что под влиянием L-аргинина происходит повышение уровня экспрессии eNOS., тогда как L-NAМЕ – снижает. L-аргинин проявляет антиоксидантные свойства, снижает интенсивность ПОЛ и стимулирует активность ферментов антиокислительной защиты клеток [4, 5]. Еще одним важным процессом, в определенной степени детерминирующим эффективность образования NO, является влияние окисленных ЛНП и лизофосфатидилхолина (продукта реакции, катализируемой фосфолипазой и ассоциированной с ЛНП) на транспорт L-аргинина в ЭК. В связи с этим положением с целью выяснения роли ЛНП в нарушении биодоступности оксида азота мы определяли в другом варианте исследований показатели обмена холестерина (ХС): концентрацию общего ХС, холестерина липопротеинов высокой плотности (ЛВП), триацилглицеридов (ТАГ) и по формуле Фридвальда рассчитывали концентрацию ХС ЛНП. Анализ данных показал статистически достоверное повышение концентрации общего ХС (р < 0,001) в сыворотке крови у крыс с экспериментальным СД. Анализ распределения ХС в липопротеинах различной плотности показал снижение его уровня в ЛВП (р < 0,001) и значительное повышение в ЛНП (р < 0,001), т.е. в атерогенных ЛП-комплексах. Одновременно происходит повышение концентрации ТАГ (р < 0,001) [5, 6].
На фоне L-аргинина происходит повышение доступности субстрата для eNOS вследствие статистически достоверного снижения в сыворотке крови ОХС и ХС ЛПН (р < 0,001), а также повышения ХС ЛВП (р < 0,001) [5]. Следовательно, в хронической стадии аллоксанового диабета имеет место гиперхолестеринемия и гипер-β-липопротеинемия, способствующие повреждению эндотелия сосудов вследствие атерогенеза, что подтверждено нами морфологически. Более того, следует предположить, что в условиях повышенного содержания в крови реактивных форм кислорода, особенно радикала гидроксила ОН– как наиболее реакционноспособного, и вторичного продукта перекисного окисления липидов (ПОЛ) – малонового диальдегида, происходит окислительная модификация ЛНП, включая переокисление липидов, образование конъюгированных диенов, углеродную модификацию (apо β), а также энзиматическое превращение фосфатидилхолина в лизофосфатидилхолин фосфолипазой. Все эти изменения, вызываемые оЛНП, способствуют нарушению транспорта L-аргинина из сыворотки крови в ЭК и уменьшению образования оксида азота и его биодоступности. Для выяснения взаимосвязи между концентрацией NO и липопротеиновым спектром сыворотки крови мы провели корреляционный анализ. Данные показали наличие отрицательной связи между содержанием NO и концентрацией ЛНП (r = –0,68). Эти результаты созвучны с литературными данными, свидетельствующими о торможении оЛНП высвобождения NO ЭК, что способствует уменьшению эндотелийзависимой вазодилатации [8]. Анализ данных микроциркуляции на фоне L-аргинина показал увеличение исходно сниженной средней и систолической скоростей кровотока, уменьшение повышенных упруго-эластических свойств сосудов микроциркуляторного русла – RI. Эти функциональные изменения являются следствием нарушенного метаболизма NO и интенсивности образования метаболитов ПОЛ, влияющих на проницаемость и тонус сосудов, угнетая эндотелийзависимую вазодилатацию, что подтверждается данными литературы [4, 5]. В противоположность этому на фоне L-NAME отмечалось нарушение микроциркуляторной гемодинамики, снижение средней и систолической скоростей кровотока и повышение периферического сосудистого сопротивления. Этому способствовало снижение концентрации NOx в сыворотке крови.
Таким образом, несмотря на достижения современной медико-биологической науки, проблема ранней диагностики специфических микроангиопатий на фоне развившейся дисфункции эндотелия сосудов и их патогенетически медикаментозная коррекция остается до конца не изученной. Выявление прогностических маркеров развития сосудистых осложнений при различных заболеваниях, в том числе и при сахарном диабете, выявление групп риска, применение различных методологических подходов к их профилактике и лечению остается в настоящее время актуальным.
Рецензенты:
Брин В.Б., д.м.н., профессор, зав. отделом физиологии и патологии висцеральных систем, ФГБУН «Институт биомедицинских исследований» Владикавказского научного центра РАН и Правительства РСО-Алания, г. Владикавказ;
Джиоев И.Г., д.м.н., профессор, зав. центральной научно-исследовательской лабораторией, гбоу впо «Северо-Осетинская государственная медицинская академия» Министерства здравоохранения Российской Федерации, г. Владикавказ.
Работа поступила в редакцию 18.02.2014.
Библиографическая ссылка
Дзугкоев С.Г., Можаева И.В., Такоева Е.А., Дзугкоева Ф.С., Маргиева О.И. МЕХАНИЗМЫ РАЗВИТИЯ ЭНДОТЕЛИАЛЬНОЙ ДИСФУНКЦИИ И ПЕРСПЕКТИВЫ КОРРЕКЦИИ // Фундаментальные исследования. – 2014. – № 4-1.
– С. 198-204;
URL: https://fundamental-research.ru/ru/article/view?id=33696 (дата обращения: 05.05.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)